【干货】一文搞懂微生物组的五大常见分析技术及应用示例
2021-03-08 09:47:10, TIANGEN
微生物组是指一个特定环境或者生态体系中全部微生物及其遗传信息的总和。其孕育着解决人类面临的包括健康下降、环境恶化等问题的关键技术和方案。
在过去的几十年里,微生物研究发展迅速,已经成为一个重大的科学和公众利益的话题。年初,Nature期刊将微生物组学选为2020年值得关注的科学之一。下面让我们跟小编一起了解一下微生物组的“家族成员”。
Q1
微生物组的概念
A:微生物组的定义,当前经常引用是Lederberg将生态环境中的微生物群落描述为生物体空间或其他环境中的共栖,共生和致病微生物群落。阅读文章时几个常见名词Microbiota, metagenome, microbiome常傻傻搞不清楚,具体差异,可见下图。

每张图说明的是同一个群体,但不同的方法可以提供不同的信息。
a. 微生物群(Microbiota):采用16S rRNA方法鉴定到的环境中微生物的种类。
b. 宏基因组(Metagenome):微生物群的基因和基因组信息,包括质粒、尤其是群体的遗传学潜能。
c. 微生物组(Microbiome):除了微生物群的基因和基因组,还包括微生物群的产物与宿主环境。
Whiteside, S., Razvi, H., Dave, S. et al. The microbiome of the urinary tract—a role beyond infection. Nat Rev Urol 12, 81–90 (2015). doi:10.1038/nrurol.2014.361
Q2
高通量测序技术在微生物组学中的应用?
A:随着测序技术的成本不断降低,相应数据分析方法也得到不断完善,为深入了解微生物组的结构和功能提供了新的手段。在分子水平上,微生物组研究分为三种层面:微生物、DNA和mRNA。相应的研究技术包括培养组(微生物基因组de novo)、扩增子、宏基因组、宏病毒组和宏转录组,各种方法的优缺点如下。

Yong-Xin Liu, Yuan Qin, Tong Chen, Meiping Lu, Xubo Qian, Xiaoxuan Guo, Yang Bai. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein&Cell. doi: 10.1007/s13238-020-00724-8
Q3
微生物基因组de novo都有哪些产品,可以得到哪些分析结果?
A:微生物基因组De novo测序分析也叫微生物基因组从头组装,指在没有该物种基因组信息的情况下,不依赖于任何参考序列信息就可对某个微生物进行分析的测序分析技术。先将细菌的染色体DNA机械地随机切割成一定长度的片段,根据不同的测序策略,构建对应大小文库,然后进行大规模测序。再用生物信息学的方法进行序列拼接获得该物种的基因组序列图谱,然后进行注释等后续一系列的分析。该分析可以预测重要基因和蛋白以了解其功能和机制,并且通过比较基因组或重测序研究进化关系。

Q4
细菌全基因组de novo测序,有没有必要做成完成图?
A:我们首先来盘点一下,细菌基因组de novo测序,做框架图和完成图的优缺点和各自的使用场景,如下图。总体而言,细菌基因组框架图成本低、获取快,完成图获得的基因组息更完整和准确。当样本量较大或者是以某些特定基因为主要分析对象时,可选择框架图。而想对某些重要的、特定的菌株进行研究或者非常看重可移动元件时,可选择完成图。

Q5
扩增子测序可得到什么结果?二代和三代全长哪个更好?
A:扩增子测序是基于特定的Marker基因或片段代表相应的物种与数据库进行比较分析,从而获得详细的物种注释信息,主要用于研究某一特定环境中微生物结构组成,及其在不同处理条件下微生物种类及丰度差异,主要优势在物种多样性层面。也可以使用PICRUSt软件通过比对16S测序数据获得的物种组成信息,推测样本中的功能基因组成,从而分析不同样本或分组之间在功能上的差异。扩增子测序主要包括16S rDNA测序、18S rDNA测序、ITS测序及目标区域扩增子测序等。
目前广泛应用的是Illumina测序技术,尽管其不断发展可以扩大其测序的通量,但其进行测序反应扩增的极限长度也不过单端300bp。因此细菌扩增子测序时,无论是选16S全长V1-V9九个区中的一个区还是两个区,我们在进行物种注释时都无法将其准确注释到物种水平而仅仅是属水平。而三代长读长的技术优势,使得16S全长测序成为可能。更长读长带来的是更加丰富的序列信息,显著提升了基于NGS测序所达不到的高水平物种识别精确度、分辨率及对样本中微生物群落的还原度。下图为基于计算机分类数据库中得到的物种分类结果。每个分支的颜色反映了每个进化枝中无法识别到物种水平的序列比例。显示几者相比,16S全长测序得到最好的物种分类结果。

Johnson, J.S., Spakowicz, D.J., Hong, B. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10, 5029 (2019). doi: 10.1038/s41467-019-13036-1
Q6
宏基因组分析可得到什么结果?
A:宏基因组学(Metagenomics)通过直接从环境样品中提取全部微生物的DNA,利用基因组学的研究策略研究环境样品所包含的全部微生物的群落组成及其功能,包括微生物多样性、种群结构、进化关系、功能活性、相互协作关系及与环境之间的关系等。
宏基因组分箱(Binning)是将宏基因组测序得到的混合了不同微生物的序列或组装得到的contigs通过核酸序列信息和序列丰度将物种分开归类的分析技术。该技术有助于获得不可培养微生物的全基因组序列,获得新物种的基因组序列和功能。TIANGEN已将Binning分析加入到宏基因组产品的标准分析列表,无需二次付费即可畅享高级分析!整体分析流程及结果如下图。

Q7
何时进行宏转录组测序?
A:宏转录组学(Metatranscriptomic)是对某一特定时期、特定环境样品中的全部微生物的RNA进行高通量测序,直接获得该环境中所有微生物转录组信息的一种测序技术的新应用,不仅可以得到微生物的物种信息,还有微生物的基因表达信息。当关注基因表达和微生物组功能活性等信息时选择该技术。
Q8
什么是宏病毒组?
A:宏病毒组(Metavirome)直接以环境中所有病毒的遗传物质为研究对象,能够快速准确的鉴定出环境中所有的病毒组成。根据研究的病毒类型,分为DNA病毒的宏组学研究和RNA病毒的宏组学研究。研究重点关注复杂样本中潜在病毒对人类及环境的危害,主要用于病毒的物种分类、相对丰度及相关功能信息研究,在病毒发现、病毒溯源、微生物预警等研究方面具有重要作用。
Q9
各种分析技术具体怎么应用?
A:微生物组研究的热潮愈演愈烈,技术的不断完善与更新,我们如何根据研究侧重,选择不同的技术。扩增子测序主要能够分析物种组成及丰度;宏基因组除了能够得到物种组成及丰度外,还可得到样本的基因注释、功能注释,进行代谢通路的研究;宏转录组是针对复杂微生物群落动态活性研究的不二选择;宏病毒组针对样本中DNA/RNA病毒类群分别进行不同病毒的病毒分类和功能注释研究。下表汇总一些相关技术的文章,可以按需进行查看。
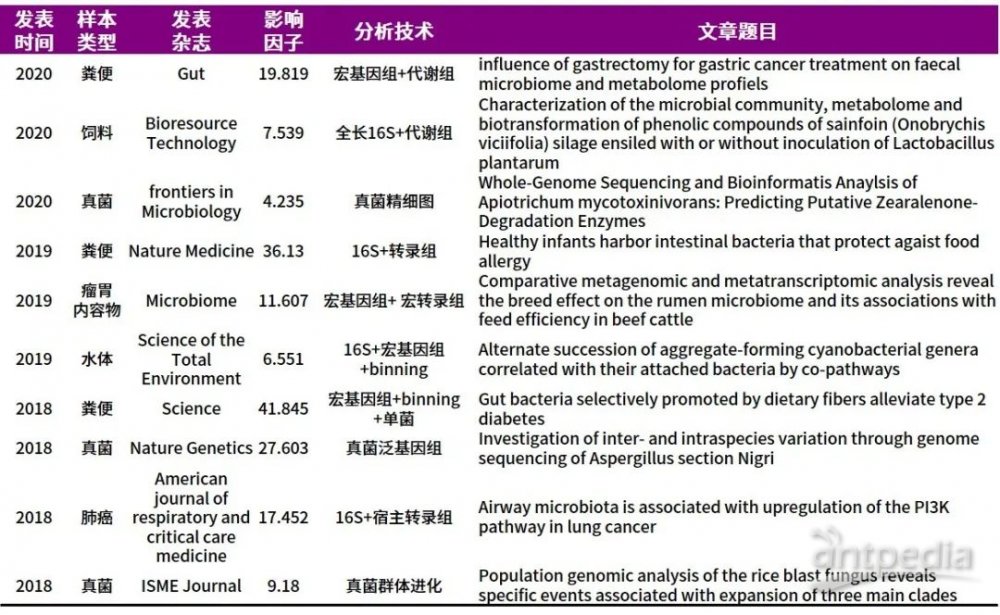
微生物组的各类分析技术特点大家get到了吗?
如果您有相关的实验及分析需求
均可联系TIANGEN~
现在联系TIANGEN
还可以参与年末促销!
签订订单最多可获4%订单金额增值金!
扫描二维码报名
还可参与在线抽奖活动!


手持云台、mini相片打印机、KFC...
![]() 等你来拿!
等你来拿! ![]()


04-17
免费试用70天!安东帕傅里叶变换红外光谱仪助力药企生产04-17 Anton Paar China
精酿人必看 | 如何让啤酒新鲜度“破局”?三大福利,抓紧来领!04-17 Anton Paar China
邀请函 | 密度黏度联合用户培训会@上海04-17 安东帕中国
脂质研究必看:2400+产品目录,四大难题一网打尽04-16
第35届化学年会圆满落幕,Sigma-Aldrich®赋能化学,共启化学探索之旅04-16
与时俱进,与药典接轨:一文看懂离子对试剂怎么选04-16 Merck
椰子水掺假?同位素质谱帮你看穿真相04-16 飞飞
纺织品PFAS风险升级,我们该如何看待“隐形污染物”?04-16 飞飞
HC57系列pH电极在陶化工艺中的应用04-16 哈希公司
5.6-5.10│海克斯康PC-DMIS测量软件初级系统培训,仅剩一个名额!04-16 卡尔希德
三坐标测量常见问题解答(三十)04-16 卡尔希德
痕量硫检测迈入新征程!磐诺自研SCD检测器,以硬核实力定义新标准04-15
案例分享 | 85%分离产率背后的“质量守护者”——expression CMS助力糖肽合成新策略04-15 汉尧
哈希旗舰店,店庆嘉年华04-15 哈希公司
H.E.L硫化物固态电池研究—BTC-130 标准电池测试绝热加速量热仪04-15 H.E.L Group
现场直击!NanoTemper 如何在2026生物制药稳定性大会实力‘破圈’?04-15 NanoTemper
谁动了我的蛋白样品?Panta帮找回被忽略的90%变质真相04-15 Melody高天歌
NanoTemper 邀您共聚第7届BIONNOVA生物医药创新者论坛@上海张江04-15 NanoTemper
客户成就 | 纳米压痕和Unity联用表征用于评估页岩的力学性质04-15 牛津仪器