80个样本就成迄今最大规模心衰蛋白质组?Circulation揭示心衰损伤机制及治疗策略!
2022-01-21 01:07:31, XHL 上海吉凯基因医学科技股份有限公司


细胞外基质(ECM)重构是心力衰竭(HF,简称心衰)的一个关键病理特征。ECM重构是持续的,并导致收缩期和舒张期损伤。全基因组关联研究表明,ADAMTS家族蛋白酶与心血管疾病有关。ADAMTS5是小鼠心脏成纤维细胞分泌的最丰富的蛋白酶之一。但ADAMTS5蛋白酶及其底物versican对心衰(HF)的贡献是未知的。
近期,伦敦国王学院的研究团队在心血管研究领域的顶刊Circulation(中科院JCR一区,影响因子29.690) 上发表了题为 “Extracellular Matrix in Heart Failure: Role of ADAMTS5 in Proteoglycan Remodeling”的文章,详细阐述了ADAMTS5在心衰细胞外基质重塑中的作用及干预方案,也是迄今最大规模的心衰蛋白质组学研究!


研究结果

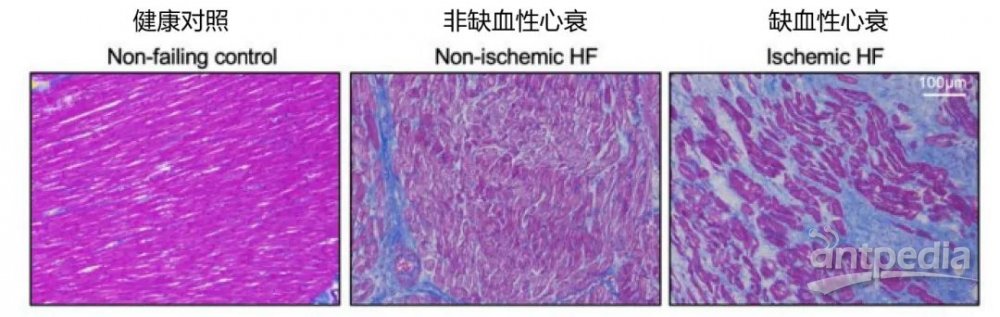
一、蛋白质组学揭示缺血性心衰心肌组织的细胞外基质(ECM)组成
(1)样本分组设计
实验组1:缺血性心衰疤痕心肌组织(n=5)
实验组2:非缺血性心衰疤痕心肌组织(n=10)
对照组:健康心肌组织(来自心脏移植供体,n=6)
和正常的健康心脏比较,非缺血性心衰存在弥漫性纤维化,缺血性心衰存在局灶性纤维化。
(2)细胞外基质(ECM)蛋白质组学结果
各组样本在提取ECM后进行蛋白质组学检测,共鉴定到200个ECM和ECM相关蛋白。缺血性心衰组织中除纤维状胶原外,间质蛋白聚糖也有积累。这些成员包括SLRP(小富亮氨酸蛋白聚糖)家族成员和主要的CSPG(硫酸软骨素蛋白聚糖)——蛋白聚糖VCAN(versican),该结果与研究者此前在猪I/R损伤的蛋白组学发现的上调VCAN类似。相比之下,在非缺血性心衰的ECM蛋白组学中,ECM蛋白的变化很少,也没有涉及到蛋白聚糖的变化。

二、ADAMTS5活性与心脏功能密切相关
ADAMTS家族的多个成员,被称为蛋白聚糖酶,可以酶切Versican。研究者此前的结果表明,ADAMTS5是小鼠分泌组中唯一鉴定到的蛋白聚糖酶。Versican被ADAMTS5酶切后可产生一个N端片段—Versikine。Versikine染色显示,其表达于小鼠心脏心肌细胞的外周区。缺血性心力衰竭患者的Versikine水平高于非缺血性心力衰竭患者。同样,在I/R损伤的猪和灌注血管紧张素II后的小鼠的心脏组织中也含有丰富的versikine。
研究者构建了ADAMTS5催化位点缺失的小鼠(Adamts5△Cat),并进行2周和4周的血管紧张素II灌注。在灌注2周和4周时,Versican在Adamts5△Cat比对照Adamts5+/+表现出显著的上升的Versican。而在基因水平上,Adamts5△Cat小鼠中血管紧张素诱导的Versican表达未有显著提升。在血管紧张素II处理后,Adamts5+/+小鼠中versikine上升,而在Adamts5△Cat小鼠中则检测不到。胶原纤维在两个基因型小鼠中相当。同时,两个基因型的小鼠在血管紧张素灌注后炎性细胞浸润水平也无显著改变。以上结果表明,ADAMTS5酶活丧失后,无法酶切Versican,导致了versican的积累,也导致了酶切产物不可检出,并影响小鼠的心脏功能受损。

ADAMTS5活性缺失会引起血管紧张素II灌注后versican的积累。Versican通过其透明质酸(HA)结合域与HA结合,是心脏中透明质酸(HA)重构的重要调节因子。研究者检测了参与心脏细胞粘附和交流的蛋白表达。血管紧张素灌注后,ITB1、FLNA和CNX43在Adamts5+/+小鼠中上调。而在Adamts5∆Cat小鼠中,这几个蛋白表达下调。因此,这些结果表明,ADAMTS5活性缺失会降低参与细胞通讯的蛋白的表达。

三、单细胞测序显示Versican和ADAMTS在人心脏中表达的特异性
研究者分析了一个已发表的人心脏单细胞测序结果,发现versican主要表达与心脏成纤维细胞,这与研究者此前小鼠心脏成纤维细胞的蛋白组学结果一致,而ADAMTS5是主要表达在心脏成纤维细胞中的ADAMTS。不同的心脏成纤维细胞群体,包括生成ECM和重塑ECM的心脏成纤维细胞,都表达高丰度的versican。ADAMTS5主要表达于一个特定的心脏成纤维细胞群体里。这个细胞群(FB1)是心室成纤维细胞的主要群体,与心房中的成纤维细胞不同。心室成纤维细胞有相对更高的ADAMTS5表达和更低的versican表达。一个左心房和左心室的蛋白组学结果也显示,在左心室中versican的表达水平更低。

四、大规模左心室蛋白质组学揭示药物处理对缺血性心衰ECM组成的影响
研究者又对更大队列的缺血性心衰的左心室样本(n=65,经过不同的药物治疗,其中44个患者使用了β受体阻滞剂治疗)进行蛋白质组学检测(分为细胞组分检测和细胞外基质(ECM)检测)。在细胞外基质的组分中共检测到177个ECM以及和ECM相关的蛋白,其中141个蛋白可被定量。使用β受体阻滞剂的样本中,发生表达变化的蛋白数量最多。GO、Reactome和KEGG分析显示了ECM合成和ECM过程的富集。而使用statins(他汀类药物)治疗的,只有很少的蛋白变化。Statins治疗显示了和代谢相关通路的富集。使用β受体阻滞剂治疗的缺血性心衰主要表现为蛋白聚糖的下调。SLRP和VCAN以及它的酶切产物versikine也受到了影响。在使用β受体阻滞剂后,HA的主要受体CD44也降低。此外,研究者还发现,心率与ECM蛋白丰度呈现正相关,其中versican蛋白丰度与心率呈现最强的相关性。而在mRNA表达水平上,并未发现显著变化。

五、去甲肾上腺素诱导心脏成纤维细胞中versican的表达
为了探究β-肾上腺素信号在心脏成纤维细胞中的作用,研究者用去甲肾上腺素处理小鼠心室成纤维细胞。TGFβ1处理作为对照。TGFβ1处理后,Adamts5表达下调。去甲肾上腺素处理后,诱导了versican的表达,而不影响Adamts5。用β1肾上腺素受体拮抗剂bisoprolol(比索洛尔)阻止了去甲肾上腺素处理心脏成纤维细胞诱导的versican表达上调。该结果表明,β受体阻滞剂在调控这个关键蛋白聚糖中的直接作用。相似地,用bisoprolol(比索洛尔)处理2周后,降低了小鼠心脏中versican的表达,而不影响Adamts5的基因表达。
六、缺血性心衰的聚类分析
基于蛋白质-蛋白质丰度相关性的聚类分析展示心衰组织细胞外基质蛋白的聚类情况。结果显示,第一个密集聚类中,Versican是这个聚类的核心,同样还有服用β受体阻滞剂导致变化的其他ECM蛋白。第二个聚类包含与非成纤维细胞的基膜相关的ECM蛋白。在主成分分析(PCA)中,临床变量没有将服用β受体阻滞剂的患者与未服用β受体阻滞剂的患者分开。但当使用细胞外基质蛋白质组学数据时,获得了良好的分离。第一主成分(PCA1),占变异的22.5%,决定了有和没有β-受体阻滞剂的患者之间的最佳分离,并且主要依赖于蛋白聚糖丰度。其他药物没有明显分离。因此,β受体阻滞剂减少心脏细胞外基质沉积,与其他临床变量无关,而蛋白聚糖受到的影响最大。

研究总结
研究者进行了80名心衰患者和6名健康(对照)的左心室组织的细胞外基质(ECM)蛋白质组学检测,是迄今最大规模的蛋白心脏ECM蛋白组学研究。先是发现了蛋白聚糖在缺血性心衰患者的心肌中积累。其次,研究者证实 ADAMTS5 在心脏重塑过程中裂解蛋白聚糖。此外,研究者证明了Adamts5活性缺失的小鼠中存在蛋白聚糖积累、心脏功能受损和参与细胞间通讯的蛋白质的减少。最后,研究者探讨了药物对细胞外基质重塑的影响,并揭示了β受体阻滞剂减弱了缺血性心衰蛋白聚糖积累。
做蛋白组学· 找吉凯
吉凯基因凭借多年在靶标筛选及验证服务领域的技术积累,建立的标准化 、工程化 、系统化的GRP平台,为中国研究型医生提供科研服务,加快科研成果转化。其中,蛋白质组学平台拥有多台timsTOF Pro、Exploris 480高精度质谱仪,专业领先的Spectronaut Plusar、Mascot等分析软件,提供专业的4D、DIA、TMT、PRM、磷酸化修饰组等检测服务,强大的机器学习算法、IPA分析、蛋白基因组分析服务,系统的生物标志物、分子分型、药物靶点、基因功能研究等解决方案,真正让广大研究型医生的科研工作更省心、更省力、更高效。




1.实验技术干货
2.蛋白质组学研究
3.腺病毒简介及应用
6.单细胞测序
8.悬浮细胞专用病毒
10.测序技术研究与应用
12.腺相关病毒选择/应用
13.表观遗传研究
14.文章解析
15.国自然课题设计思路解析
16.生物信息分析及工具
17.外泌体研究
18.肿瘤免疫研究
19.高分文章

05-08 圈内人都会关注
节庆喧嚣渐远,在皖仪“劳动与奉献”赞歌正亮!05-07 WAYEAL皖仪
这不是玄学,来试试不挑质谱的高深度血浆蛋白质组学!05-07 莲莲看
一文读懂|全球首款自复制RNA(saRNA)疫苗ARCT-154背后的专利05-07 耀海生物
【聚焦光谱】请看:一机多用,服务于两个不同光学实验!05-07 聚焦光电前沿领域
网络讲座复播视频:原子力显微镜缺陷检测的相关应用05-07 Park原子力显微镜
食品安全国家标准 | 食品中氯酸盐和高氯酸盐的的测定05-07 安捷伦科技
解构生物大分子 | 安捷伦与您相约全国生物医药色谱质谱及相关技术学术交流会05-07 安捷伦科技
【液体吸光度测量】典型配置、硬件说明、软件操作05-07
【会议预告】杭州大微与您相约第五届乳制品检测与控制技术交流会05-06 DW
【获奖名单公布】高纯度细胞色素C纯化工艺方案05-06 分离纯化部
珂睿邀您共同关注食品安全国家新标准方法解读!速来预约→05-06
展会邀请|天瑞仪器邀您莅临2024 CPCA SHOW国际电子电路(上海)展览会05-06
新芝生物2024渠道商交流会首站(杭州)圆满成功05-06 SCIENTZ
报名开始丨 EDS&EBSD高级应用培训班(北京站)05-06
QbD理念在工艺和杂质控制策略制定中的应用05-06 ACD/Labs
【百特小课堂】什么叫标准物质的不确定度05-06
中国第一,全球第五!丹东百特上榜“世界工业新闻”年度报告05-06
全国最大的电池展在重庆开幕,丹东百特豪华阵容参展05-06 丹东百特
BT-1001智能粉体特性测试仪05-06