【First-in-class药设系列】靶向G1-S检查点受损肿瘤的Cyclin A/B RxL抑制剂设计
2025-09-29 04:08:46, 秦慧 & 张健 TargetMol中国
小细胞肺癌(Small-cell lung cancers, SCLC)是一种具有高度侵袭性且治疗手段有限的肺癌亚型。其典型基因组特征包括RB1和TP53基因的功能丧失性突变,导致G1–S期检查点功能失调,进而引起E2F转录因子(early region 2 binding factors, E2F)的异常激活。类似地,在其他癌症类型中,G1–S检查点也常因CDKN2A缺失、Cyclin D/E扩增等事件而受损,同样会导致E2F信号高度活化,促进肿瘤细胞增殖。值得注意的是,尽管E2F活性在细胞周期进程中不可或缺,其过度激活反而可诱导细胞凋亡。
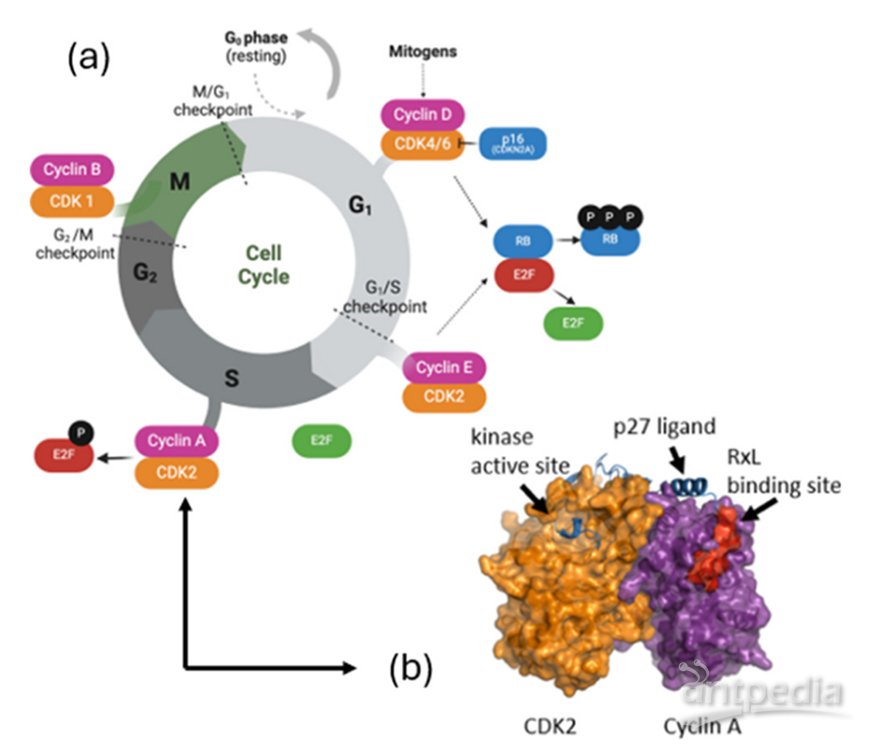
Cyclin A–CDK2复合物能够通过识别E2F1蛋白中的RxL基序(Arg-x-Leu motif)并使其磷酸化,从而负向调控E2F1的转录活性。然而,由于该蛋白-蛋白相互作用界面较宽且结构相对平坦,靶向该结合位点的传统小分子抑制剂开发面临较大挑战。近日,来自哈佛医学院的研究人员开发一类可透膜的套索肽抑制剂,用以特异性阻断Cyclin A–CDK2与E2F1之间的相互作用,解除其对E2F1的负调控作用,从而进一步加剧E2F信号的异常激活,最终选择性地诱导pRB通路失调和E2F过度活化的癌细胞发生凋亡。

他们基于已发表的Cyclin A2–CDK2复合物与结合沟抑制剂的晶体结构(PDB: IURC),以及该复合物与激酶抑制蛋白 p27Kip1 的晶体结构(PDB: IJSU),通过叠合这两个配体中的RxL基序,设计了一种新型套索肽骨架(序列为:Lys-Pro-Ser-Ala-Cys-Arg-Lys-Leu-Phe-Gly),旨在通过同时占据Cyclin蛋白表面的疏水区域(hydrophobic patch, HP)及相邻的次要疏水口袋(secondary hydrophobic pocket, S)提高其与Cyclin蛋白的亲和力。文章的总体思路是基于此骨架模板,利用Circle pharm的合成与计算平台对初始骨架进行N-甲基化、环大小调整和亲脂性优化等化学修饰,显著提高了化合物的透膜性。通过生化实验鉴定出具有亲和力的特征基团,并将这些基团融合到渗透性骨架上,以实现效价、透膜性和理化性质的平衡。

通过荧光偏振(fluorescence polarization, FP)和表面等离子共振(Surface Plasmon Resonance, SPR),他们检测化合物对Cyclin A、B、E的结合力与选择性;借助Circle Pharma公司的合成与计算平台,采用N-甲基化、调整环大小、优化亲脂性等常用策略提高化合物的被动细胞渗透性;同时同步优化溶解度、LogD等类药性参数。其中,先导化合物CIRc-004表现出优异的特性,它可平衡地抑制Cyclin A(IC50 = 0.13 μM)和Cyclin B(IC50 < 0.02 μM),而对Cyclin E的抑制较弱(IC50 = 11.6 μM),在体外展现出抗增殖活性。令人意外的是,研究发现单独抑制Cyclin A或Cyclin B的CIRc-0018、CIRc-019效果均不理想,CIRc-004的非活性对映体则完全没有活性,这意味着必须同时抑制两者才能高效诱导细胞死亡。

随后,为了验证这类环肽抑制剂可选择性杀伤E2F高活性的细胞,他们对46株SCLC细胞进行了筛选,发现几乎所有上述细胞均对低浓度的CIRc-004敏感。基因集富集分析(Gene set enrichment, GSEA)和变异分析(Gene set variation, GSVA)显示,CIRc-004药物敏感性与E2F靶标基因及G2/M检查点通路基因的表达呈强相关性,这种相关性超越了SCLC的分子亚型(如ASCL1、NEUROD1、POU2F3或YAP1亚型)。提示无论肿瘤属于哪种转录谱系,只要其E2F信号高度活跃,就对药物敏感。这些结果表明E2F活性是一个比传统SCLC分型更优越的预测指标。
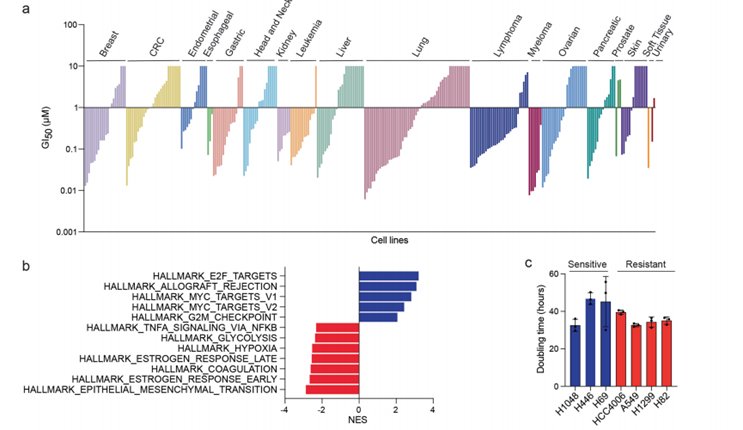
为确定更广泛的适用性并确保Cyclin A/B RxL抑制剂不具有普遍毒性,他们使用CIRc-001(广谱Cyclin A/B/E RxL抑制剂)对302种癌细胞系进行了筛选。约180种细胞系(包括肺、乳腺和卵巢模型)显示出对CIRc-001的选择性敏感性(EC50 < 1µM)。E2F靶区基因特征在各类癌症的敏感细胞系中富集,而上皮-间质转化相关特征则在耐药细胞系中富集。因此,低E2F活性和高表达上皮-间质转化相关基因可能预示对Cyclin A/B RxL抑制剂的耐药性。

他们进一步比较了CIRc-004在敏感SCLC(NCI-H1048、H446和H69)细胞系和耐药非小细胞肺癌(NSCLC)细胞系(A549、HCC4006和H1299)中的反应。结果表明,细胞对CIRc-004的敏感性并非由增殖速率差异导致,且非活性对映体CIRc-005对所有测试细胞系均无效。在敏感SCLC中,CIRc-004诱导了细胞凋亡和有丝分裂阻滞。这些效应需要同时抑制Cyclin A和Cyclin B,因为Cyclin A抑制剂CIRc-018和Cyclin B抑制剂CIRc-019并不会影响细胞的增殖、凋亡和细胞周期。此外,正常细胞(包括RPE1和CD34+造血干细胞及祖细胞)对CIRc-004不敏感,即使在高剂量下也未出现有丝分裂阻滞或凋亡,提示该药物具备较宽的治疗窗口。相反,CIRc-004在微摩尔水平诱导了p53表达和G1期阻滞。最后,具有较低E2F特征的SCLC细胞系NCI-H82也对CIRc-004耐药。综上,高E2F活性的癌症患者最可能从这种Cyclin A/B RxL抑制剂中受益。

通过全基因组CRISPR-Cas9筛选,他们揭示了Cyclin A/B RxL抑制剂诱导细胞凋亡依赖于纺锤体组装检查点(spindle assembly checkpoint, SAC)的完整激活。在敏感细胞系NCI-H1048中,筛选结果表明,敲除SAC核心组分(如KNTC1、MAD1L1、ZWILCH等)或细胞周期蛋白B1基因(CCNB1)本身可赋予细胞对CIRc-004的耐药性,而敲除转录抑制因子E2F7/E2F8则增强敏感性。这一发现提示Cyclin B-CDK2复合物的功能获得性激活是引发SAC依赖性细胞死亡的关键机制。遗传验证实验证实,CCNB1或SAC基因(如KNTC1、MAD1L1)的敲除可有效挽救CIRc-004引起的凋亡和有丝分裂阻滞。此外,使用MPS1抑制剂化学破坏SAC功能或表达显性负性CDC20(R445Q)突变体均可拮抗CIRc-004的致死效应,进一步确认SAC通路的激活是这类抑制剂发挥抗癌作用的必要前提。

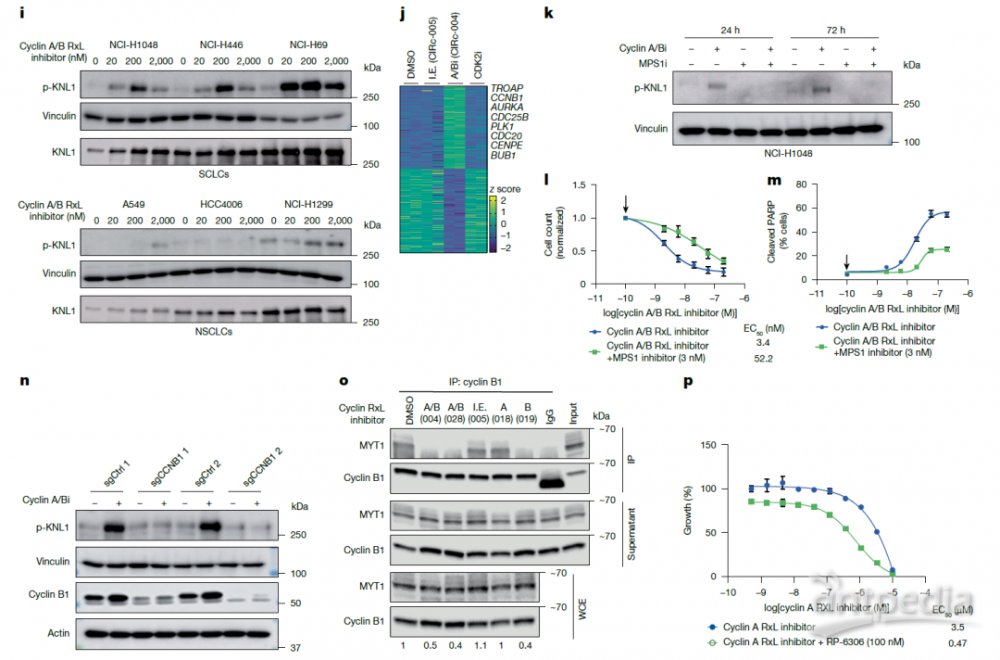
在机制探索中,研究人员发现Cyclin A/B RxL抑制剂通过阻断Cyclin B与激酶MYT1之间的相互作用从而激活SAC。Co-IP实验证实,双功能抑制剂CIRc-004及Cyclin B选择性抑制剂CIRc-019可有效破坏Cyclin B与MYT1的结合,而Cyclin A选择性抑制剂CIRc-018或无活性对映体CIRc-005则无此效应。由于MYT1是CDK1的负调控因子,该相互作用的破坏导致Cyclin B–CDK1活性升高,表现为Cyclin B Ser126位点磷酸化增强。更重要的是,在CCNB1敲除的细胞中,CIRc-004无法诱导SAC标志物p-KNL1的产生,表明Cyclin B的活性是SAC激活所必需的。为验证该通路的功能性贡献,他们联合使用Cyclin A选择性抑制剂CIRc-018与MYT1小分子抑制剂RP-6306,发现这一联合用药可在多个SCLC细胞系中有效诱导细胞死亡;反之,联合使用负调节CDK1活性的WEE1抑制剂则无此效果, 这些结果一致表明, Cyclin A/B RxL抑制剂的致死效应特异性依赖于破坏cyclin B–MYT1轴,解除了对CDK1的抑制,促进了SAC的过度激活和有丝分裂灾难(mitotic catastrophe, MC),而非通过Cyclin B-WEE1相互作用通路。


随后,本研究揭示了Cyclin A/B RxL抑制剂通过驱动形成一种非经典的Cyclin B-CDK2复合物来诱导细胞死亡的核心机制。CRISPR-Cas9筛选发现,位于Cyclin B与CDK1/2相互作用界面的残基(如Glu169、Tyr170、Tyr177)发生突变后可导致对CIRc-004的耐药,提示该界面对于药效至关重要。进一步的Cyclin B免疫沉淀-质谱分析证实,CIRc-004处理显著减少了其与天然调控因子MYT1、p27和p57的结合,但意外地富集了CDK2。尽管Cyclin B在细胞内通常与CDK1结合,但Co-IP实验验证,CIRc-004处理特异性增强了内源性Cyclin B与CDK2的相互作用,而不影响其与CDK1的结合。这种诱导形成的Cyclin B-CDK2复合物具有功能活性,可磷酸化其非经典底物Stathmin(STMN1),进而促进SAC的激活。功能必要性实验表明,CDK2的缺失可部分减弱CIRc-004诱导的SAC激活;而在nocodazole阻滞后释放的细胞中,CDK2化学抑制剂则能完全阻断CIRc-004的SAC激活效应。这些数据共同阐明,Cyclin A/B RxL抑制剂通过重新连接细胞周期网络,促使Cyclin B与CDK2形成一种功能获得性的促凋亡复合物,该复合物通过磷酸化一系列下游底物(如Stathmin),导致纺锤体检查点超活化并最终驱动有丝分裂细胞死亡。

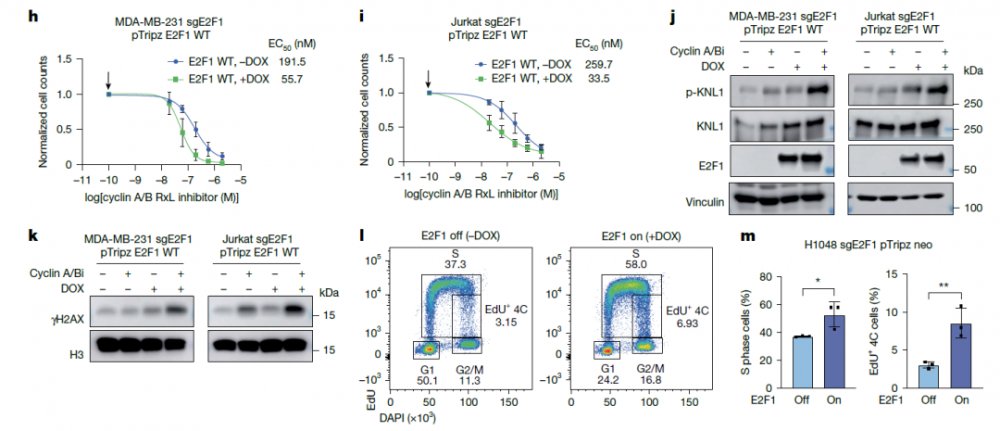
他们发现CIRc-004处理使敏感SCLC细胞大量停滞在DNA复制的S期晚期/G2期,揭示了该药物通过扰乱DNA复制进程并阻止细胞完成分裂。这种E2F的超活化在敏感细胞中引发了严重的复制压力,表现为DNA损伤标志物γH2AX及复制压力信号p-RPA2(Ser33)和p-KAP1(Ser824)的显著积累。Co-IP实验证实,Cyclin A/B双重抑制剂(CIRc-004, CIRc-028)及Cyclin A选择性抑制剂(CIRc-018)可有效破坏Cyclin A与E2F1/E2F3的相互作用,阻断了Cyclin A-CDK2介导的E2F负反馈调控环路,致使E2F驱动转录程序异常增强。功能上,E2F超活化是药物发挥杀伤效应的必要条件:在NCI-H1048、MDA-MB-231及Jurkat细胞中,通过多西环素(Dox)诱导E2F1(或E2F2/E2F3)的过表达,可显著增强细胞对CIRc-004的敏感性,并伴随更强的DNA损伤反应和SAC激活。值得注意的是,尽管E2F1过表达或CDK2抑制均能模拟CIRc-004诱导的S期/G2期DNA持续合成现象,但二者单独均不足以引发有丝分裂阻滞和细胞死亡。这一发现凸显了Cyclin A抑制(导致E2F超活化与复制压力)与Cyclin B抑制(导致SAC超活化)之间的不可或缺的协同作用,共同构成了诱导合成致死的完整机制。

为验证Cyclin A/B RxL抑制剂的抗肿瘤活性,研究者通过药代动力学优化获得环肽CIRc-028。其在NCI-H69和NCI-H1048移植瘤模型中经静脉注射(100 mg/kg,连续14天)可显著抑制肿瘤生长且耐受良好,肿瘤组织免疫组化(IHC)结果显示瘤内p-KNL1与cleaved caspase-3表达升高,表明其通过激活SAC及诱导凋亡起效。在两种化疗耐药SCLC PDX模型(DFCI-393/402)中,口服环肽分子CIRc-014同样表现出持续抑瘤效果与良好安全性。药效学及药代动力学分析证实,治疗后瘤内SAC与凋亡标志显著上升,药物暴露量达有效浓度,且转录组富集G2/M及有丝分裂相关信号,一致表明其体内作用具靶向性与机制特异性。综上,Cyclin A/B RxL抑制剂在代表不同疾病背景的体内模型中均表现出强劲的抗肿瘤活性,其作用由靶点驱动的SAC激活和凋亡所介导,并拥有良好的安全性和药代动力学特性,为其向临床转化提供了强有力的依据。
综上,本文从SCLC最根本的生物学缺陷出发,通过理性药物设计攻克了PPI靶点,首次开发了一类可口服的Cyclin A/B RxL环肽抑制剂,并深入阐释了其独特的“合成致死”机制,为RB1和TP53缺失的癌症患者带来了新的希望。目前,基于该研究开发的化合物CID-078已经进入I期临床试验。
参考文献
1. Singh, S., Gleason, C.E., Fang, M. et al. Targeting G1–S-checkpoint-compromised cancers with cyclin A/B RxL inhibitors. Nature (2025). https://doi.org/10.1038/s41586-025-09433-w
2. Bockus AT, Leung SSF, Fraga-Walton B, et al. Discovery of Cell-Permeable Macrocyclic Cyclin A/B RxL Inhibitors that Demonstrate Antitumor Activity. J Med Chem. 2025;68(16):17030-17045. doi:10.1021/acs.jmedchem.5c00253
04-14 安捷伦科技
不容小觑!复杂宠物食品基质中多类别真菌毒素检测04-14 安捷伦科技
药物分析排忧解难系列 | 中药检验时间紧任务重,究竟该咋办04-14 安捷伦科技
助力嫦娥探月,安捷伦串联无机质谱破解月球身世密码04-14 安捷伦科技
婴幼儿奶粉维生素 K1 异构体分析-安捷伦国产柱 ValueLab GP‑C3004-14 安捷伦科技
“老友”记 | 寻找安捷伦 6890 金牌守护者,一键解锁专属福利04-14 安捷伦科技
开班通知|真机实操,真材实料,纽迈分析2026第一届核磁应用培训班硬核启航04-13 纽迈分析
缘起南开聚滨海 共探纯化新高度 —— 南开大学生物医药企业家校友考察团走进博蕴生物04-13
准直光束秒变贝塞尔光束?高精度圆锥透镜(Axicons)选型指南04-13 韵翔光电
液相色谱柱内径,你真的选对了吗?04-13 技尔 GL Sciences
心相聚·岳向前|博鹭腾2026年合作伙伴大会圆满落幕04-13
活动预告 | 德国斯派克将参加中国国际数据中心液冷关键技术年会04-13 市场部
纳克微束邀您参会|2026年华东地区电子显微学学术交流会04-13 纳克微束
纽迈展风采 | 聚焦第二届地球能源国际前沿论坛:纽迈以低场核磁技术赋能绿色勘探04-12 纽迈分析
【直播预告】肿瘤免疫课题攻坚难!组织孵烂了,关键免疫 marker 还不显色……04-12
纽迈展风采|微观透视,智创未来——纽迈分析携低场核磁共振技术亮相2026军民两用新材料论坛04-11 纽迈分析
CMEF 2026看点:《GE医疗》版04-10
直播预告 | 类器官应用介绍04-10
徕卡显微系统在中国正式发布 Viventis SCAPE ——亮相中国细胞生物学大会04-10 徕卡显微系统
显微课堂 | 线虫研究指南-针对线虫的相关工作04-10 徕卡显微系统