反相色谱肽分离色谱柱和流动相研究第二部分:色谱柱表征方案的耐用性评估
2024-04-28 13:50:22, 阎作伟 编译 Advanced Chemistry Development, Inc. (ACD/Labs)
本篇文章已经开通留言功能,
欢迎至文末留言交流讨论!
反相色谱肽分离色谱柱和流动相研究
第二部分:色谱柱表征方案的耐用性评估

本文使用简化因子(Reduced Factor Design)设计评估肽反相色谱(RPC)色谱柱表征方案的稳健性,以确定所需控制的程度。评估包括包括温度、流速、梯度滞留体积、梯度的变化、甲酸体系水相和有机相中的甲酸含量、甲酸铵体系的pH值和强洗脱中乙腈的量(%MeCN), 发现其中 MeCN 的量的变化会导致不可接受的结果。为确保数据的完整性,引入了缓解措施,令使用肽探针对 反相色谱柱表征方案最终落地,并描述了最终方案。此外,本文还评估了仪器和色谱柱批次间的差异性,令实验具有良好的重现性。
01介绍
选择合适的固定相是方法开发的关键因素,然而,由于许多不同的色谱柱制造商提供了大量的固定相,因此很难对从哪个色谱柱开始做出合理的决定。对于小分子方法开发,有多种策略,包括田中Tanaka方案及Tanaka扩展方案、施耐德Snyder的疏水减法模型和莱塞利尔Lesellier的线性减法能量关系(LSER),它们都表征了反相(RP)固定相,其结果可在数据库中免费访问[1–5]。这些数据库令最终用户能够确定用于方法开发目的的各种色谱柱的差异性,或者选择色谱表现相似的色谱柱,以选择“备用”色谱柱。
直到最近,对于适用于多肽分离的RP色谱柱,还没有相应的表征方案,而本系列文章提出的多肽RPC色谱柱表征方案就是为了解决这一缺陷而开发的[6]。本研究合成了26种具有不同物理化学性质的专门设计的肽,以评估突出的相互作用,如疏水性、氢键、静电相互作用和芳香作用特性,并解决典型降解途径的相关问题。使用了 14 根色谱柱对这 26 种肽进行评估,这些色谱柱相的选择基于对色谱柱化学的先验知识。在低和中等 pH 值下使用梯度色谱法进行了评估。使用归一化保留差异 (Δtg ∗) 测量选择性,其中总共产生了 66 个 delta 值来探测不同类型的相互作用,这些相互作用使用主成分分析 (PCA) 进行了严格评估。
选择的流动相是甲酸(pH 2.5)和甲酸铵(pH 6.45,天然pH),以评估固定相的更纯净的相互作用。流动相选择的基本原理可以参考文献[6]。固定相被分为三个不同的组,以描述突出的相互作用;中性(具有高度配体密度和/或封端的色谱柱)、负电性(在配体中含有带负电荷的部分,由于配体键合密度低/封端率低而能够发生亲硅相互作用的色谱柱,或能够形成极性相互作用如氢键)和正电性(包含带正电荷的部分的色谱柱)。
所需的肽数量从26个减少到11个,产生11个Δ值,以描述正电荷/负电荷、疏水性、酚类特性、芳香族特性以及立体结构、外消旋、脱酰胺和氧化的变化(表1和表2)。因为TFA经常用于肽分析,还进行了一项比较的研究,以观察TFA代替甲酸作为低pH值添加剂的效果,之后放弃了TFA的使用。由此产生的数据比对甲酸/甲酸铵Bi-plot发现失去了类别区分力,这表明在使用TFA时会丢失信息。因为TFA具有离子对效应,固定相的特性因此不那么重要,由此可见使用适当的添加剂(如甲酸)进行色谱柱表征方案的合理性。此外,进行了一项迭代研究,评估了 66 个 delta 值,以确定是否可以减少研究数量,同时仍然充分描述色谱柱的选择性。

表1和表2
表1和表2 在Kinetex Evo C18上用标准方法测量11种肽以及其Delta 值,tg以及tg*
为了使多肽RPC色谱柱表征方案真正有价值,必须考虑表征方案的稳健性[7]。ICH指南将稳健性定义为“衡量其不受方法参数微小但故意变化影响的能力,并表明其在正常使用期间的可靠性”[8,9]。该方案的稳健性是使用通常称为DoE(实验设计)的简化因子设计来评估,这是学术界和制药行业评估稳健性最常用的工具之一。在这种方法中,探索了反映与该方案相关的预期实验误差的微小系统变化[9-11]。可以使用替代方法,例如一次一个因子(OFAT),每次改变一个变量,保持所有其他感兴趣的参数不变。然而,它通常被证明是更耗费资源(即时间、材料等),并且可能无法估计不同变量之间的相互作用。从DoE中可以得出大量具有统计学意义的信息,而这些信息无法通过OFAT方法获得[12]。11个肽Δ值被用作创建DoE模型的响应,该模型突出了偏离程度,从标称中心点(即原始方法条件)产生的系统变化。这些数据表明哪些变量具有统计学意义,以及它们与结果的实际相关性,从而突出了必须仔细控制哪些操作参数,以保持稳健的方法。在评估更多的色谱柱以建立一个数据库之前,必须考虑程序的稳健性。因为这些固定相将被包含在色谱柱表征数据库中,以便个人合理地选择用于肽/蛋白质分离的固定相。为了确保色谱柱数据库的完整性,使用了类似的方法来评估Tanaka和扩展方案的稳健性[10]。
本文旨在更好地了解可能影响方法稳健性的关键因素,这不仅包括对方案中使用的液相色谱参数执行DoE,还包括确定流动相pH切换和样品负载的稳健性。最后,将确定方案中使用的液相色谱参数的实际操作限值,以保持基于肽的色谱柱表征数据库完整性的可接受的可重复性和可靠性。本文是系列文章的一部分,旨在研究影响肽分离系统的因素,其中首先评估固定相,然后研究不同流动相添加剂的效果,以帮助色谱工作者更合理地制定自己的分离研究计划。
02实验
2.1. 化学品,试剂和仪器
除非另有说明,否则所有液相色谱分离均在岛津Nexera X2 UHPLC系统上进行,其模块之前已描述[6]。每种肽的碱基序列位于表2中,参考文献[6]中已描述了所有肽探针。
2.2.因子设计

表3
上文中描述了多肽RPC色谱柱表征方案条件。表3中列出了每种特定色谱柱上样测量的混合物。每种测试混合物都含有计算精确 delta 值所需的两种肽。这有时意味着在多个测试混合物中存在肽的重复。这是为了确保对 delta 值进行良好描述并提高方法的稳健性,因为微小程度的保留漂移可能会影响细微的相互作用,例如外消旋。所选择的肽基于参考文献[6]中进行的迭代消除研究,既包含细微的变化也包含显著的变化,评估常见的降解和特定的相互作用,描述用于表征的色谱柱。delta 值是梯度洗脱中选择性的量度,用作 DoE 的输入。
表4
表4总结了DoE中评估的因素,包括不同的±1水平。所研究的因素被选为最可能的误差来源,其中确定的水平反映了人们可以预期的随机变化(即至少是预期标准差的 3 倍)。基于仪器规格和鉴定数据的误差传播计算被用于标准偏差的估计[13]。使用Modde Pro软件(见第2.4节)创建和评估简化因子设计,该设计使用八个实验,不同变量具有不同的+1或-1水平,以及对中心标称条件设定了三个重复测试,以确定程序的可重复性(表5和表6为甲酸和甲酸铵梯度)。针对标称条件生产不同批次的溶剂。该软件创建了一个随机运行顺序,以消除结果中的任何偏差。

表5表6
2.3.仪器的差异
另外三种液相色谱仪器为Waters H-Class, Waters I-Class (Milford, Ma, USA) 以及Agilent 1290 二元系统(Waldbronn, Germany),使用相同批次的流动相和固定相。三种仪器的梯度滞留体积在 300 到 700µ L 之间 . 沃特世仪器由 Empower 3(Feature Release 3)控制,而安捷伦由 OpenLab CDS(Chemstation C.01.07 SR4)控制。
2.4.固定相
多肽RPC色谱柱表征方案使用14种不同的固定相[6] 150×2.1 mm的标准化色谱柱规格开发的。所有色谱柱均为制造商提供的新色谱柱。使用Phenomenex Kinetex Evo C18(150×2.1 mm,100Å,2.6 μm)进行简化因子设计实验,同时在Waters Acquity HSS C18(150×2.1 mm,130Å,1.8 μm)上考察仪器变化。在6个不同批次的Ascentis Express C18上评估了色谱柱批次间的差异性(150×2.1 mm,90Å,2.7 μm)。水峰顶点被用作每根色谱柱的死时间标记[14]。
2.5.软件和计算
主成分分析(PCA)使用SIMCA (Version 14.1, Umetrics, Umeå, Sweden) 和Origin (Version Origin Pro 2016, Origin Lab, Northampton, MA, USA)完成. PCA 中的变量都是自动缩放,以便使每个变量具有相同的重要性。使用Modde Pro(版本12.0.1)进行因子设计。在pH 2.5和6.45下,使用通用蛋白质/质量分析(GPMAW)软件 (Version 9.51, Lighthouse Data, Odense, Denmark).计算肽探针的净电荷。
03
结果和讨论
3.1.主成分分析和因子设计
用一阶多项式模型拟合甲酸和甲酸铵设计的数据(表5和表6)来评估每个delta值(表4)的稳健性。在中心点条件下,在Kinetex Evo C18上获得的典型Δtg和Δtg *值如表1所示。模型的质量使用拟合模型的回归系数(R2)和模型交叉验证的回归系数(Q2)来衡量[11,15]。对于一个好的模型,两个值都接近 1。然而,在稳健性评估中,理想的结果是一个较差的模型,即低 R2 和 Q2 值。对于稳健的方法,不同因子的 ±1 变化的影响应与标称条件下的实验噪声相对应。甲酸体系中Δ值的平均R2和Q2分别为0.761(标准偏差SD 0.208)和-0.133(标准偏差SD 0.151),而甲酸铵Δ值的平均R2和Q2分别为0.750(SD 0.080)和-0.200(SD 0.000)。中心点重复实验(N9-11)为程序的可重复性提供了指示,其中delta探针的平均差异为0.001(相关delta值的范围为0.000至0.004)。该结果表明多肽RPC色谱柱表征方案是稳健的。可以通过比较系数图来进一步评估方法的稳健性,该系数图评估每个参数对稳健性的单独影响(图 1 和图 2)。

图1 图2
每个变量都按比例缩放并在系数图中居中,以便它们具有可比性。条形图的高度给出了影响的程度,而误差条表示 95% 置信区间,当误差线不为零时,该区间突出显示了具有统计意义的参数。
3.1.1.温度
温度对甲酸铵和甲酸中的大多数反应没有明显的影响,但是,甲酸中的Δ(8a,1)和Δ(16,13)有统计学反应。然而,柱线的高度表明,这两个增量结果的实际意义微乎其微。虽然对于这些探针肽的影响很小,但建议确定色谱柱的实际温度,因为众所周知,柱温箱的设计甚至色谱柱在柱温箱内的位置会改变色谱柱的实际温度。可以通过使用预混的MeCN/H2O流动相(45:55 w/w)检测苯丙酮(250 nm检测波长),以消除仪器间%MeCN的变化。柱温箱温度在 30–60 ◦C 范围内以 5 ◦C 的间隔变化。然后,在色谱柱进行恒温之前,将同一色谱柱浸入装有 30 cm 0.12 mm 管的水浴中,以测试同一色谱柱。然后假设这是对色谱柱温度的准确测量。水浴的温度应使用具有适当精度的校准/认证温度计确定,例如± 0.5 ◦C 之间的 30-60 ◦C。样品在30至60◦C下以5◦C的间隔进样,以构建图(保留时间-系统死时间)与温度的关系图,以确定LC系统的任何温度偏差(即在特定保留时间下的ΔT)。任何新型柱温箱设计都应遵循此程序,一旦确定了色谱柱的实际温度,就可以调整肽RPC色谱柱表征方案,以补偿温度的任何偏差。
3.1.2.梯度组成的变化
甲酸和甲酸铵的响应均不受梯度系统变化的影响。梯度的这种系统偏移假设在梯度长度上应用相同程度的误差。
3.1.3. 流速
流速的变化也对应于梯度斜率的变化。流速对甲酸中的Δ(9,1)仅表现出很小的统计学响应,而甲酸铵和甲酸中的所有其他响应均不受影响。与温度类似,该变量的实际影响非常小,因此可以假设该参数在方法中是稳健的。
3.1.4. 滞留体积
对于任一流动相中的所有响应,驻留体积在统计学上均不显著,表明该变量不会影响甲酸或甲酸铵梯度的稳健性。这在很大程度上是由于保留时间的归一化,消除了停留体积的影响,允许在不同仪器之间直接比较。选择用于评估的停留体积范围 (100–500 μL) 应涵盖 UHPLC 仪器。
3.1.5.甲酸的含量
甲酸水平的差异可能会影响方案的稳健性,因为不同的水平会导致不同的pH值,从而影响肽上的整体净电荷。然而,观察到甲酸体积的差异不显著,Δ(9,1)和(16,13)值表现出非常小的统计学意义,但被认为没有实际意义。但建议从预先校准的移液器中按体积分配甲酸体积,每次制备溶剂时都要检查移液器,以确保色谱结果的完整性。
3.1.6. 甲酸铵储备溶液pH值
由于残留硅醇的pKa值范围,中等pH值下的固定相环境有些不可预测[16,17]。据信,大多数硅醇应在pH 6.45(甲酸铵的天然pH值)下电离,然而,这种不确定性可能导致结果出现更大程度的变化,因此可能导致缺乏稳定性。甲酸铵也可能是误差的来源,其中研究了缓冲液的配置时间、储存环境和由此产生的pH范围。测量了 16 种不同的 200 mM 溶液的 pH 值,其中平均 pH 值为 6.45 (SD 0.03)。DoE中的水平(pH 6.45±0.06)是根据基于pH值的三次测定的99%CI设定的。甲酸铵的配置时长似乎对溶液的整体pH值没有太大影响,其中测得的pH值在DoE测试的范围内。然而,基于储存不当时,pH值会发生变化。原因是盖子不好的容器的甲酸铵溶液pH值较低,这表明氨发生了损失。这可能会影响硅醇电离程度,从而影响保留率和Δ值。理想情况下,甲酸盐应储存在干燥器中以减少吸水量,并降低氨损失的风险。
甲酸铵的响应均稳定在DoE的pH上限和下限内,无统计学意义。应使用适当校准的标准品测量储备缓冲溶液的pH值,以确保pH值在此范围内,以确保方案的完整性。还建议,如果甲酸铵除了 pH 值变化外还表现出任何相当大的吸湿性迹象,则不应使用。为避免微生物生长,从而可能污染液相色谱系统并可能堵塞色谱柱入口筛板,导致峰裂和背压升高,建议将储备缓冲溶液的储存时间限制在 5 ◦C 下 时长不超过4 个月。
3.1.7.有机相的乙腈含量
Δ (3,1)、Δ (9,1)、Δ (10,9) 和 Δ (24,13) 的选择性特别容易受到甲酸铵梯度中 B 溶剂中乙腈含量的变化的影响。与之前的变量不同,这种影响实际上足够显着,具有实际意义,因此值得进一步研究。正如之前定义肽RPC色谱柱表征方案的文献中所述,PCA用于在Bi-plot图中可视化Δ值和色谱柱之间的异同[3,18]。甲酸铵和甲酸DoE的delta值被包含在PCA中,以评估该方法对方案的稳健性,区分不同的固定相的能力,并查看所提出的MeCN水平对结果完整性的限制。从生成的双图(图3)中可以看出,除了位于左下象限下方的一小部分运行外,大多数DoE运行都聚集在原点附近。进一步的评估表明,B溶剂中MeCN含量的-1水平使该子集被拉走了。

图3
在一段时间内,流动相储层中的溶剂蒸发可能会造成MeCN的损失。因此,我们进行了蒸发研究,以确定液相色谱溶剂盖可以合理损失的物质。将安捷伦阀盖(Waldbronn, Germany)和 SCAT 安全盖(Mörfelden-Walldorf, Germany)与用于溶剂储存的封闭盖进行了比较。在30天内,封闭瓶盖的重量减轻了0.00%,这表明乙腈在储存过程中没有损失,但是,据计算,Agilent和SCAT瓶盖每天的损失分别为0.04%和0.03%,这实际上可能是有问题的。
为了解决这个问题,采用的方法将 B 溶剂从 MeCN/H2O (80:20 w/w) 中的 20 mM 甲酸铵改为 100% MeCN。相应地调整梯度以获得相同的MeCN体积分数,并与原始方法进行比较,具有相似的色谱结果,而不管B溶剂中的缓冲液浓度如何降低(在梯度中肽通常洗脱的位置,甲酸铵浓度从18 mM降低到12 mM)。
3.2.仪器差异
在不同的液相色谱仪器之间成功转换液相色谱方法的能力对于该方案的广泛接受极为重要。这在梯度色谱中尤为重要,因为色谱柱体积和驻留体积的贡献已被证明会导致相当大的选择性差异[19]。
将三种肽混合物进样到Acquity HSS C18色谱柱上,分别使用三种不同的仪器(Agilent 1290、Waters Acquity H-和I-class),梯度滞留体积范围在300至700 μL之间,大于DoE中评估的范围。在所有比较中都使用了相同批次的流动相和色谱柱,以消除因流动相制备差异而产生的任何差异,并了解仪器对研究的贡献。记录每种仪器的 delta 值并进行比较,然后通过 PCA 分析数据并置于Bi-plot图中(图 3)。
这三种仪器都在 95% 的置信限圈内。这种变化与DoE的预期相当,因此表明在不同类型的仪器上获得的结果应该是可比的。
3.3.色谱柱批间差异
使用六根Ascentis Express C18色谱柱评估色谱柱批次间的差异性。其中三根色谱柱含有相同批次的硅烷,其余色谱柱使用了另外三批硅烷。所有批次间色谱柱均使用新方案进行测试,减少探针数量(即去除易受MeCN变化影响的探针)和流动相,以消除它们对任何变异性的影响。批次间的观察结果可以在bi-plot图(图4)内被圈起来,看到的分散是由于批次间的变化。本实验是在同一天使用相同的仪器和流动相收集的,其对结果的影响已被消除。结果也与之前使用各种方案对其他色谱柱进行的批间研究一致[9,20–26]。这突出表明,Bi-plot之间的任何偏差都是由色谱柱的选择性差异引起的,因此使用这种方法区分色谱相似或不同的固定相是可行的。

图4 色谱柱批间差异
3.4.缓慢的平衡
Snyder等很早就发现某些反相固定相在中等pH值变为低pH值时平衡速度缓慢。它表现为在改变流动相pH值时保留时间会发生稳定漂移。据信,在所有市售的色谱柱中,约有40%表现出某种形式的平衡缓慢[27]。这种现象的确切机制尚不清楚,但据推测,随着具有低表面电荷的现代硅胶的出现,pH 值的变化可能需要很长时间才能重新平衡,这表现为可电离物质的保留漂移。使用肽作为探针评估一系列C18型色谱柱时,该方案同时使用低pH值和中等pH值。使用甲酸梯度条件在C18固定相上重复进样肽测试混合物,结果一致[图5(A)]。然后将该相暴露于甲酸铵梯度条件下,在重复进样中,肽混合物快速平衡,表明从低到中间条件时可以获得一致的结果。然而,当同一色谱柱再次暴露于甲酸梯度时,它未能产生与暴露于中间pH值之前的结果相当的结果(保留时间缓慢降低 - 见图5(B))。所有峰的保留时间都有所增加,但进样之间的保留时间持续减少。文献表明,静态平衡可以恢复平衡缓慢的色谱柱[27-29],然而,即使在低pH值条件下进行过夜静态平衡,也无法将该固定相恢复到其原始色谱保留率[图5(C)]。
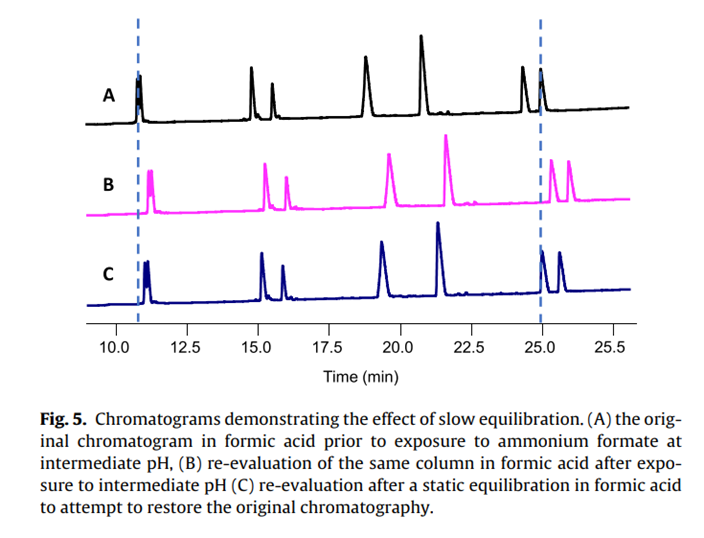
图5
这种现象不仅局限于甲酸,而是可电离物质从中等或高pH值到低pH值的任何转换之时都会发生。使用TFA也无法纠正这个问题,这种添加剂的离子对效应意味着需要专用色谱柱,这对于色谱柱表征是不切实际的[30]。尽管已经设计了一些固定相来应对这种缓慢的平衡问题,例如Acquity CSH系列相[31],但仍有许多市售色谱柱确实表现出这种现象。因此,决定首先使用甲酸梯度表征每个固定相,然后使用甲酸铵在中间pH值下进行测试,以避免任何有害的保留漂移或需要过长的平衡时间。
3.5.载样量
色谱效率极易受到分析物过载的影响,这会导致峰形不佳,尤其是对于可电离物质。为避免色谱性能受到影响,肽和蛋白质分离的允许载量通常要低得多[31]。关于载样量以及合理化过载效应,有大量文献可用[32-34]。
使用DMSO/H 2 O(80:20 v / v)对 [L-Asp3]-Bovine GLP-2(1-15)的储备溶液(1mg/mL)进行连续稀释。使用甲酸梯度色谱条件将每种溶液重复地注入Kinetex Evo C18(150×2.1 mm,2.6 m)色谱柱。甲酸的低离子强度会产生最坏的过载情况;因此,选择它来观察载样量和过载的影响。

图6
总共进行了八次稀释(为简单起见,图6中显示了四次),并叠加了所得色谱图。亲水肽在pH 2.5时的净电荷为+1.2,在酸性条件下,出现的典型的直角前延峰和极端拖尾,随着负载的增加,显示出特征性的“鲨鱼鳍”峰形状。随着色谱柱负载的增加色谱峰保留时间减少,因此会影响用计算Delta值的归一化保留时间。每种肽发生这种效应的程度可能不同,因此必须很好地描述每种肽的载荷,以保持一致的 delta 值。本研究中,峰形不是关键问题,因为保留时间必须一致,因此必须保持负载恒定。在设计色谱方法时,选择能够提供更好图6的流动相至关重要。使用甲酸梯度条件,不同负载(0.031-2.000 g)的亲水肽[D-Asp3]-牛GLP-2(1-15)的叠加峰分布。该峰表现出特征性的Langmuir等温线,随着样品负载的增加,峰拖尾显著。生物制药行业通常使用基于磷酸盐的系统来获得好的峰形。
根据所使用的固定相类型,负载曲线也可能不同。例如,对Acquity CSH系列的固定相进行了优化,为肽类等提供了更好的峰形和效率,以提供线性等温线,而不是典型的Langmuir等温线(图6)。与慢速平衡效应类似,过载行为被认为是由于表面电荷的变化,其中CSH范围的平衡表面电荷抵消了该问题,从而产生对称、有效的峰[31]。
当涉及到样品负载时,样品溶解度也至关重要。在pH 2.5和6.5时,亲水肽的净电荷分别为+1.1至+1.2和-4.7至-3.7。疏水肽在 pH 2.5 时的净电荷为 +2.2,而在 pH 6.5 时的净电荷为 0。电荷为0可能会发生溶解度问题以及入口熔块沉淀和堵塞色谱柱。长时间暴露于中间pH条件后,观察到压力增加和色谱柱性能下降。更换入口筛板和扫描电子显微镜证明颗粒已沉积在筛板上。因此,为了尽量减少这种情况发生的可能性,减少了多肽的负载,本研究并在进样器和色谱柱之间安装了在线过滤器。
3.6. 缓解措施:调整肽反相色谱柱表征方案
简化因子设计的理想情况是发现所评估的参数在统计上或实际上都不相关。但是,如果存在实际相关的参数,则必须采取缓解措施以减少影响。
甲酸和甲酸铵DoE的梯度系统变化、流速和梯度滞留体积以及甲酸铵的pH值的公差均为可接受。尽管被认为具有统计学意义,但两个DoE的温度限值以及水溶液和有机物中的甲酸百分比的实际相关性可以忽略不计,因此使用甲酸表征的方法在规定的限值内可以被认为是稳健的。然而,乙腈在甲酸铵梯度的B溶剂中的浓度已被证明是具有统计学意义的结果,具有实际意义。浓度的降低对六个 delta 值中的四个产生了影响,导致敏感的 Δ(3,1)、Δ(9,1) 和 Δ(10,9) 探针从表征中去除了。去除的Δ值是外消旋的探针,在甲酸中仍由Δ(9,1)和Δ(14,13)表示。此外,在没有受影响探针的情况下,还评估了score plot的完整性。
据信,稳健性实际上将比本研究中显示的要好得多,因为引入的缓解措施(表7)将进一步减少变异。更新的肽RPC色谱柱表征方案在附录I中进行了描述。

表7 增强方法耐用性的手段和原理
04结论
本文使用简化的因子设计和 PCA 评估肽 反相色谱柱表征方案的稳健性,并系统地改变各种因素以推断对方案细微变化的影响。结果表明,甲酸梯度可以看出,在本研究的给定公差范围内提供了可靠的结果。然而,甲酸铵梯度需要缓解以提高乙腈在B相中浓度的稳健性。评估的所有其他参数均不影响稳健性。还确定了色谱柱的样品负载量,并评估了某些市售固定相对低pH值和中pH值之间切换的潜在影响。这两项研究都对方案产生了影响,并采取了缓解措施来解决这两种现象。此外,还评估了三种不同液相色谱配置的仪器变异性和色谱柱间批次间的变异性,以确定可预期的变异程度。液相色谱和色谱柱的变异性都很小,这突出表明,使用肽RPC色谱柱表征方案观察到的固定相之间的差异是由色谱选择性差异引起的,而不是随机误差。
为了提高稳健性,提出了一些修改建议,因此,展望未来,结果应该提供比本研究中显示的更高的可靠性和可重复性。这将为使用肽作为探针表征不同的固定相并区分其选择性差异提供更大的信心,从而允许选择互补的固定相进行方法开发或为备用方法选择类似的色谱柱。
附录1


参考文献:
向上滑动阅览
[1] ACD Column Selection Database, 2018 (Accessed: 23/10/2018) https://www. acdlabs.com/resources/freeware/colsel/.
[2] U.S. Pharmacopeial Convention, 2018 (Accessed: 23/10/2018) http://apps.usp. org/app/USPNF/columnsDB.html.
[3] M.R. Euerby, P. Petersson, Chromatographic classification and comparison of commercially available reversed-phase liquid chromatographic columns using principal component analysis, J. Chromatogr. A 994 (2003) 13–36.
[4] N.S. Wilson, M.D. Nelson, J.W. Dolan, L.R. Snyder, R.G. Wolcott, P.W. Carr, Column selectivity in reversed-phase liquid chromatography I. A general quantitative relationship, J. Chromatogr. A 961 (2002) 171–193.
[5] C. West, E. Lemasson, S. Bertin, P. Hennig, E. Lesellier, An improved classification of stationary phases for ultra-high performance supercritical fluid chromatography, J. Chromatogr. A 1440 (2016) 212–228.
[6] J.K. Field, M.R. Euerby, P. Petersson, J. Lau, H. Thørsen, Investigation into reversed-phase chromatography peptide separation systems Part I: development of a chromatographic protocol for column characterisation, J. Chromatogr. A (2018), http://dx.doi.org/10.1016/j.chroma.2019.05.038.
[7] B. Dejaegher, Y.V. Heyden, Experimental designs and their recent advances in set-up, data interpretation and analytical applications, J. Chromatogr. A 1158 (2007) 158–167.
[8] M.E. Swartz, I. Krull, Method validation and robustness, LCGC North America 24 (2006) 480–490.
[9] Guidelines for Industry, Text on Validation of Analytical Procedures, 2005, ICH-Q2A, November.
[10] P. Petersson, M.R. Euerby, An evaluation of the robustness of the Tanaka characterization protocol for reversed-phase liquid chromatography columns, J. Sep. Sci. 28 (2005) 2120–2129.
[11] G.E.P. Box, J.S. Hunter, W.G. Hunter, Statistics for Experimenters: Design, Innovation, and Discovery, 2nd ed., Wiley, New Jersey, 2005.
[12] V. Czitrom, One-factor-at-a-Time versus designed experiments, Am. Stat. 53 (1999) 126–131.
[13] J.C. Miller, J.N. Miller, Statistics for Analytical Chemistry, 3rd ed., Ellis Horwood Limited, Chichester, 1993.
[14] P. Petersson, B.O. Boateng, J.K. Field, M.R. Euerby, A practical approach to modelling of reversed-phase liquid chromatographic separations: advantages, principles and possible pitfalls, LCGC Europe 31 (2018) 120–143. 112 J.K. Field et al. / J. Chromatogr. A 1603 (2019) 102–112
[15] D.C. Montgomery, E.A. Peck, G.G. Vining, Introduction to Linear Regression Analysis, 5th ed., Wiley & Sons, Inc, New Jersey, 2012.
[16] Uwe D Neue HPLC Columns: Theory, Technology and Practice, 1st ed., Wiley-VCH, Inc, 1997.
[17] A. Méndez, E. Bosch, M. Rosés, U.D. Neue, Comparison of the acidity of residual silanol groups in several liquid chromatography columns, J. Chromatogr. A 986 (2003) 33–44.
[18] I.T. Jolliffe, Principal Component Analysis, Springer, Berlin, 2011.
[19] P. Petersson, M.R. Euerby, M.A. James, Translations between differing liquid chromatography formats: advantages, principles and possible pitfalls, LCGC North America 32 (2014) 558–567.
[20] M. Kele, G. Guiochon, Repeatability and reproducibility of retention data and band profiles on reversed-phase liquid chromatography columns: I. Experimental protocol, J. Chromatogr. A 830 (1999) 41–54.
[21] M. Kele, G. Guichon, Repeatability and reproducibility of retention data and band profiles on reversed-phase liquid chromatographic columns: II. Results obtained with Symmetry C18 columns, J. Chromatogr. A 830 (1999) 55–79.
[22] M. Kele, G. Guichon, Repeatability and reproducibility of retention data and band profiles on reversed-phase liquid chromatography columns: III. Results obtained with Kromasil C18 columns, J. Chromatogr. A 855 (1999) 423–453.
[23] M. Kele, G. Guichon, Repeatability and reproducibility of retention data and band profiles on reversed-phase liquid chromatography columns: IV. Results obtained with Luna C18 (2) columns, J. Chromatogr. A 869 (2000) 181–209.
[24] M. Kele, G. Guichon, Repeatability and reproducibility of retention data and band profiles on reversed-phase liquid chromatography columns: IV. Results obtained with Vydac 218TP C18 columns, J. Chromatogr. A 913 (2001) 89–112.
[25] M. Kele, G. Guichon, Repeatability and reproducibility of retention data and band profiles on six batches of monolithic columns: IV. Results obtained with Luna C18 (2) columns, J. Chromatogr. A 960 (2002) 19–49.
[26] U.D. Neue, C.H. Phoebe, K. Tran, Y.F. Cheng, Z. Lu, Dependence of reversed-phase retention of ionizable analytes on pH, concentration of organic solvent and silanol activity, J. Chromatogr. A 925 (2001) 49–67.
[27] D.H. Marchand, L.A. Williams, J.W. Dolan, L.R. Snyder, Slow equilibration of reversed-phase columns for the separation of ionized solutes, J. Chromatogr. A 1015 (2003) 53–64.
[28] D.V. McCalley, Overload for ionized solutes in reversed-phase high-performance liquid chromatography, Anal. Chem. 78 (2006) 2532–2538.
[29] D.V. McCalley, Study of overloading of basic drugs and peptide in reversed-phase high-performance liquid chromatography using pH adjustment of weak acid mobile phases suitable for mass spectrometry, J. Chromatogr. A 1075 (2005) 57–64.
[30] J.W. Dolan, Ion pairing – blessing or curse? LCGC Europe 21 (2008) 258–263.
[31] P.C. Iraneta, K.D. Wyndham, D.R. McCabe, T.H. Walter, A Review of Waters Hybrid Particle Technology. Part 3. Charged Surface Hybrid (CSH) Technology and Its Uses in Liquid Chromatography, 2011 (Accessed: 25/10/2018) http:// www.waters.com/webassets/cms/library/docs/720003929en.pdf.
[32] S.M.C. Buckenmaier, M.R. Euerby, D.V. McCalley, Overloading study of bases using polymeric RP-HPLC columns as an aid to rationalization of overloading on silica-ODS phases, Anal. Chem. 74 (2002) 4672–4681.
[33] M.M. Fallas, S.M.C. Buckenmaier, D.V. McCalley, A comparison of overload behaviour for some sub 2 µm totally porous and sub 3 µm shell particle columns with ionised solutes, J. Chromatogr. A 1235 (2012) 49–59.
[34] G.B. Cox, L.R. Snyder, Preparative high-performance liquid chromatography under isocratic conditions: III. The consequences of two adjacent bands having unequal column capacities, J. Chromatogr. A 483 (1989) 95–110.
[35] M.M. Fallas, M. Hadley, D.V. McCalley, Practical assessment of frictional heating effects and thermostat design on the performance of conventional (3µm and 5 µm) columns in reversed-phase high -performance liquid chromatography, J. Chromatogr. A 1216 (2009) 3961–3969.
[36] R.G. Wolcott, J.W. Dolan, L.R. Snyder, S.R. Bakalyar, M.A. Arnold, A.J. Nichols, Control of column temperature in reversed-phase liquid chromatography, J. Chromatogr. A 869 (2000) 211-230
本篇文章为反相色谱肽分离系统研究第二部分,本研究共有四个部分,将陆续发出,敬请期待!
前期回顾:
反相色谱肽分离系统研究第一部分:开发色谱柱表征方案
ACD/Labs CN
微信号|ACDLabsCN
05-24
实战演练,技能精进——连华科技助力郑州市生态环境执法大队技能培训05-24
5月18-28日,快检系列双重优惠火热进行中!05-24
【海能实验室】膳食纤维测定仪测定可溶性、不可溶性及总膳食纤维含量05-24
陕西、北京、天津、湖北……渠道商交流会再更新!05-24
佳绩频传,宸安生物5月再获三项殊荣!05-24 市场战略中心
Opentrons Flex:让文库制备流程更便捷!05-24 Opentrons
2024半导体先进技术创新发展和机遇大会圆满闭幕05-24 优尼康
展会预告丨2024中国平坦化技术大会05-24 优尼康
【展会预告】钢研纳克邀您参加CISILE 第21届科仪展05-24
「钢研纳克」即将在 WCNDT 2024世界无损检测大会闪亮登场05-24
闪耀制造之巅|钢研纳克获颁制造业单项冠军企业证书05-24
HPLC-ELSD分离检测鲸蜡硬脂醇聚醚-2505-24 Unimicro
胤煌YinHuang与您相约武汉丨2024药品质量控制与检验技术大会05-23
上海交大/上硅所黄富强课题组设计新型超快充钠离子电池负极材料 | 用户成果速递05-23 HORIBA
【设备更新仪器推荐】高速高分辨显微共焦拉曼光谱仪——LabRAM Odyssey05-23 HORIBA
2024 ASMS,快来看看有哪些亮点?05-23 沃特世
半导体行业解决方案之共聚物分析05-23 沃特世
[inform]2024沃特世信息学全球用户大会圆满落幕!05-23 沃特世
UPLC,20岁生日快乐!05-23 沃特世