基于分子网络与MS-DIAL及自建数据库快速发现橙花瑞香中具稀有骨架的愈创木烷型倍半萜类化合物
2025-12-09 15:04:13, CHEMICO Advanced Chemistry Development, Inc. (ACD/Labs)


在分子网络、MS-DIAL及自建数据库联用策略的指导下,从橙花瑞香(Daphne aurantiaca)中靶向分离得到11个新型愈创木烷型倍萜(1–11)及4个已知类似物(12–15)。其中,dapurant A–B(1–2)为罕见的愈创木烷型倍萜二聚体,由两种不同环骨架的愈创木烷单元构成;dapurant C–D(3–4)为稀有的氯化愈创木烷型倍萜。
通过综合运用以下方法明确了化合物的结构与立体构型:
核磁共振波谱分析
计算机辅助结构解析(ACD/Structure Elucidator)
密度泛函理论核磁计算(结合定制化DP4+分析、CP3分析、MAEΔΔδ偏差评估)
DU8+手性识别
电子捕获检测器计算
Rh2(OCOCF3)4诱导电子圆二色谱分析
半合成验证
网络药理学分析提示并验证了该类化合物在体外与计算模拟中具有乙酰胆碱酯酶抑制活性及抗Aβ蛋白聚集活性。活性评估结果凸显了化合物1、13与14作为新型神经系统药物先导结构的开发前景。
具有新颖骨架的天然产物在植物源中通常含量极低,仅依靠传统的重复柱色谱技术难以从复杂混合物中快速分离此类化合物。这种现状催生了促进天然产物研究的创新策略,使科研人员能够以更高效、务实的方式开展工作1。
分子网络作为天然产物发现中最常用的策略之一,在可视化及注释复杂质谱数据的结构关联方面发挥着关键作用。在我们先前对药用植物的研究中,基于MS/MS的分子网络已成功分离出一系列具有独特结构和显著活性的化合物2−7。然而,该策略的局限性亦不容忽视。
尽管全球天然产物社会分子网络(GNPS)库已收录超过70,000张MS/MS谱图,但其对已知化学空间的覆盖仍存在局限。在分子网络中,仅少数聚类或节点能够被注释,这不利于实现数据的信息可视化与去重复化。
为解决化合物鉴定与注释的瓶颈,我们探索了一种名为MS-DIAL的开源软件流程。该流程适用于非靶向代谢组学研究,可兼容数据依赖性与前体离子依赖的MS/MS碎裂方法。与现有其他软件不同,MS-DIAL整合了四类信息源:精确质量、同位素分布、保留时间预测及MS/MS碎片匹配8。更重要的是,它既可调用公共数据库谱库,也支持用户构建自定义数据库,从而有效避免假阳性结果的产生。
瑞香属(Daphne)植物广泛分布于亚洲至欧洲地区,是瑞香科(Thymelaeaceae)中物种多样性最丰富的属9−11。本课题组长期致力于从该属植物中发现结构独特且活性显著的化合物,前期研究表明倍半萜类成分是该属主要的生物活性物质1,2,12−24。
为更高效地发掘瑞香属中具有新颖结构和重要生物活性的倍半萜类成分,我们系统整理了该属已分离倍半萜化合物的二级质谱数据,构建了专属数据库。本文首次将分子网络技术、MS-DIAL分析平台与自建数据库进行策略性整合,对尚未充分研究的橙花瑞香(Daphne aurantiaca)开展新型萜类成分发掘,成功获得11个具多样环骨架的愈创木烷型倍半萜新化合物。以下将详细阐述目标化合物的分离鉴定、结构解析与生物活性评价工作。
对橙花瑞香(Daphne aurantiaca)的C4−1至C4−7组分进行液相色谱-质谱联用(LC-MS/MS)分析,以优先筛选结构新颖的倍半萜类成分。通过GNPS平台的分子网络工作流处理数据,将相似谱图聚类为可视化分子家族。为筛选与倍半萜相关的分子家族并预测目标成分的极性范围,联合应用MS-DIAL工作流与自建数据库。经MS-DIAL的MS/MS谱库匹配,在两个分子家族中共注释出7个节点为愈创木烷型倍半萜(图1)。

图1. 基于MS-DIAL技术注释的橙花瑞香(D. aurantiaca)C4-3、C4-5、C4-6和C4-7组分分子网络图
分子网络中相邻节点通常具有高度相似的MS/MS谱图,因此与上述7个节点相关联的节点均为潜在目标。根据GNPS分子网络信息与MS-DIAL分析结果,溯源目标节点对应组分的高效液相色谱(HPLC)保留时间,最终靶向分离获得11个新型愈创木烷型倍萜(1−11)及4个已知化合物(12−15)(图2)。

图2. 化合物1–15的化学结构
化合物1(dapurant A)为黄色油状物,其(+)-HRESIMS显示m/z 503.2765 [M+Na]+离子峰(计算值C30H40O5Na, 503.2768),确定分子式为C30H40O5,不饱和度为11。
1H NMR谱中可见:
3个烯烃质子信号δH 4.71(单峰, 1H)、4.73(单峰, 1H)和6.07(四重峰, 1H, J=1.5 Hz)
5个次甲基信号δH 2.05(多重峰, 1H)、2.26(多重峰, 1H)、2.52(多重峰, 1H)、2.63(重叠峰, 1H)和3.01(双二重峰, 1H, J=10.5, 2.5 Hz)
5个甲基信号δH 0.65(二重峰, 3H, J=7.2 Hz)、1.02(二重峰, 3H, J=7.2 Hz)、1.69(单峰, 3H)、1.74(单峰, 3H)和1.99(二重峰, 3H, J=1.5 Hz)
13C NMR谱显示30个碳信号,包括:
5个甲基
6个次甲基
9个亚甲基
10个季碳
其中低场区可识别:
3个羰基碳信号δC 203.0、202.3和172.5
6个烯碳信号δC 172.1、168.0、150.5、139.6、127.7和109.4
上述波谱特征表明化合物1为二聚倍半萜结构。
通过¹H-¹H COSY相关信号(图S2)识别出三个独立的自旋耦合系统:
H-4/H-5/H2-6/H3-15
H-11/H2-13
H-10′/H3-14′
HMBC相关信号(图S2)进一步验证了四个结构片段的存在:
H-5与C-10的³J相关
H-9与C-7的³J相关
H-11与C-1和C-12的³J相关
H2-13与C-2和C-12的³J相关
H3-14与C-1和C-9的³J相关
H3-15与C-5的³J相关
7-OH与C-6和C-8的³J相关
H2-8′与C-11′的³J相关
H3-13′与C-7′和C-12′的³J相关
H3-14′与C-1′和C-9′的³J相关
H3-15′与C-3′和C-5′的³J相关
由于部分位点核磁信号重叠,直接连接片段存在困难(图3)。

图3. ACD/Structure Elucidator与核磁计算联用确定化合物1平面结构的工作流程示意图
采用CASE系统基于经验性核磁化学位移预测生成可能结构列表,并通过¹³C平均偏差值(dA(¹³C)、dN(¹³C)、dN(¹³C+¹H))排序。排名第一的候选结构(ID=1)因缺乏H2-2′与C-6′、H2-6′与C-2′的³J HMBC相关信号而被排除。
结合瑞香属倍萜的结构特征,ID=2的预测结构更合理(图3)。进一步采用GIAO方法进行核磁化学位移计算,确认1为罕见的愈创木烷型二聚体,由两个不同环骨架的愈创木烷单元构成。

图S1. 采用计算机辅助结构解析(CASE)专家系统生成的化合物 1可能结构
以已知化合物daphnauranol D和wikstromone C为起始原料,通过两步半合成法制备化合物1(实验部分),确证其整体结构(如下图所示)。

Scheme 1. 化合物1与2的合成路线
计算与实验ECD谱图高度吻合,最终确定其绝对构型为(1R,4S,5S,7S,11S,1′R,7′R,10′S)(图S5)。
HRESIMS分析确定分子式为C30H40O5(m/z 503.2766 [M+Na]+,计算值503.2768)。核磁数据表明2与1结构相似,但存在以下显著差异:
H-2′(δH 2.78, 2.44)
H-10′(δH 1.69)
C-1′(δC 88.2)
C-2′(δC 46.2)
C-10′(δC 47.8)
提示2为1的C1′差向异构体。通过半合成与量子化学计算验证其绝对构型为(1R,4S,5S,7S,11S,1′S,7′R,10′S)(Scheme 1,图S5)。
分子组成经(+)-HRESIMS离子峰分析确定为C15H23ClO2。对化合物3和4的一维(1D)与二维(2D)核磁谱图的详细解析表明,二者为C-11差向异构体。
NOESY谱中观测到的交叉峰支持:
H-1与H-7呈α取向
H3-13与H3-14呈β取向(图S3)
由于C-7与C-11间的碳-碳单键可自由旋转,无法通过NOESY交叉峰确定C-11的相对构型。
因此,通过计算两个C-11差向异构体的核磁化学位移,并选用CP3结合MAEΔΔδ作为关键参数,评估计算与实验数据的匹配度27,28。
鉴于重原子的特性,常规核磁计算方法通常不适用于卤代天然产物(图4)。本研究采用快速精准的DU8+方法,计算含重原子碳的13C核磁化学位移29。

图4. 基于计算与实验核磁化学位移的化合物3和4的CP3计算(a)与MAEΔΔδ参数分析(b)
较低的MAEΔΔδ值表明:
构型A与化合物3的匹配度最优
构型B与化合物4的匹配度最优
且CP3计算结果显示1S,7R,10S,11R构型(A)归属于化合物3的概率为100%,由此确定了3和4的相对构型。
基于空间位阻规则,化合物3和4的Rh2(OCOCF3)4诱导CD谱在350 nm附近观测到的Cotton效应,分别指认了C-11的R与S构型30(图5)。实验与计算ECD数据的一致性进一步验证了3和4的绝对构型(图S5)。

图5. 化合物3(a)和4(b)的Rh-ICD光谱及Gerards-Snatzke体积效应对应规则
Dapurant E (5) 的分子式经HRESIMS数据确认为C15H20O3。一维核磁谱图显示其含有三个骨架甲基、四个亚甲基、两个次甲基和六个季碳(包括两个酮羰基和四个烯碳),表明该化合物为倍半萜类。
¹H-¹H COSY相关信号(H2-2/H2-3/H-4/H-5/H2-6/H3-15)证实了C-2至C-6的连续连接关系。HMBC相关信号(CH2-12与C-8、CH3-13与C-7和C-12、CH3-14与C-1和C-9)提示存在两个α,β-不饱和羰基。关键HMBC相关(H2-2与C-10、H2-3与C-1、H2-6与C-8和C-11)进一步连接了上述结构片段,从而确定5为8,9-开环-8,12-愈创木内酯,其平面结构如图2所示。
由于手性中心质子的NOESY相关无法确定相对构型(图S3),对两种可能异构体进行GIAO一维核磁计算。较高的R²值(0.9985)和100%自定义DP4+概率表明(4R,5R)-5a为最可靠结构(图S4)。(4R,5R)-5a的计算ECD谱与实验谱高度吻合,由此精确确定了5的绝对构型(图S5)。
Dapurant F (6) 为黄色油状物,HRESIMS数据推断其分子式为C14H18O。与phaeocaulisin R的谱学数据对比表明,6为phaeocaulisin R的去羟基类似物31。HMBC相关(H3-15与C-3、C-4和C-5)验证了该结构。实验ECD谱与计算谱一致,表明6的绝对构型为4S(图S5)。
Dapurant G (7) 的分子式经HRESIMS确定为C15H22O3(m/z 251.1635 [M + H]+,C15H23O3计算值251.1642),不饱和度为5。一维及二维核磁谱分析显示其具有三环片段(5/6/7稠合环系),含一个α,β-不饱和羰基(δC 126.1, 170.6, 204.4)。
关键¹H-¹H COSY相关(H-2/H-3/H-4/H-5、H-4/H3-15、H-12/H-11/H3-13)与HMBC相关(H2-2与C-10、H2-3与C-1、H3-14与C-1、H-9与C-1/C-7、H-11与C-6、7-OH与C-6/C-8、H2-6与C-1)共同确证了7的平面结构(图S2)。
NOESY相关(H-5与H3-14、Hβ-6与H2-13/H3-15、H-9与H-11)指认了其相对构型(图S3)。基于计算与实验ECD谱比对,确定其绝对构型为1R,4S,5S,7S,11S(图S5)。
Dapurant H (8) 的HRESIMS显示m/z 303.1573 ([M + Na]+,C16H24O4Na计算值303.1572),分子式为C16H24O4。¹³C{¹H} NMR与DEPT实验解析出16个碳信号:1个酮羰基碳(δC 206.3)、2个烯碳(δC 167.5, 139.0)、1个含氧季碳(δC 79.8)、5个次甲基(δC 109.8, 81.2, 42.8, 39.1, 39.0)、3个亚甲基(δC 50.3, 28.4, 21.4)、3个甲基(δC 13.7, 10.5, 8.7)及1个甲氧基(δC 54.0),五个不饱和度中两个由双键贡献,表明8为三环化合物。
基于¹H-¹H COSY相关,勾勒出两个自旋耦合系统:H-6/H-7/H2-8/H2-9/H-11/H3-13与H-10/H3-14(粗键标示)。HMBC相关(H2-2与C-4/C-5、H-6与C-4、H-7与C-5、H-12与C-6、CH3-13与C-12、CH3-14与C-1/C-9、CH3-15与C-3/C-5)揭示8具有6,12-愈创烷型骨架(图S2)。C-1位羟基与C-12位甲氧基分别通过OH-1与C-5、OCH3-12与C-12的HMBC相关确认。由此确立8的平面结构。
NOESY相关(OH-1与H-7、H-6与H3-13/H3-14、H-12与H3-13)指认其相对构型(图S3)。计算ECD谱与实验CD谱一致,确定绝对构型为(1R,6S,7S,10S,11R,12R)-8(图S5)。
Dapurant I (9) 为白色无定形粉末,其HRESIMS数据表明分子式为C16H24O3,不饱和度为5。对比化合物8与9的¹H和¹³C{¹H} NMR数据,发现二者主体结构基本一致,唯一区别在于C-1位取代基不同(8为–OH,9为–H),该差异通过C-1化学位移及¹H-¹H COSY与HMBC关键相关信号得以确认(图S2)。NOESY数据证实9的相对构型与8相同(图S3),其绝对构型通过二者高度相似的ECD曲线确定(图S5)。
Dapurant J (10) 与 dapurant K (11) 为一对差向异构体,分子式均为C16H20O4。经HSQC与DEPT实验分析,对比其与9的一维核磁数据,发现结构高度相似。与9不同之处在于10和11存在α,β-不饱和γ-内酯环,该结构通过二维核磁数据深入分析得以确认。此外,H3-13与C-12的HMBC相关指认了10和11的12-酮结构,而OCH3与C-6的相关信号表明甲氧基连接于C-6位。由此确立10与11的相同平面结构(图S2),并通过NOESY数据确定二者为C-6位异构体(图S3)。将四种可能构型的计算ECD曲线与实测谱图比对,确定化合物10与11的绝对构型分别为1S,6R,10S与1S,6S,10S(图S5)。

已知化合物经文献比对鉴定为:daphnauranol D (12)26、daphnauranol C (13)32、(1R,4S,5S,7R)-12-去甲瑞香烷-9-烯-8,11-二酮 (14)33 和 auranticanol A (15)34。本课题组聚焦于发掘瑞香属中具有重要生物活性的成分,发现其特征性倍萜类成分可能具有神经保护作用6,12-19。
因此,通过网络药理学方法分析了化合物1–15抗阿尔茨海默病(AD)的相关网络生物通讯预测。京都基因与基因组百科全书(KEGG)通路富集分析与GO富集分析提示,需进一步研究这些化合物对胆碱酯酶的抑制活性及抗Aβ聚集活性(图6)35-41。
结果显示:
化合物1具有显著抗Aβ聚集活性(IC50 = 12.607 ± 0.215 μM)
化合物13与14对丁酰胆碱酯酶(BuChE)的抑制呈剂量依赖性,IC50值分别为4.096 ± 0.108 μM与2.919 ± 0.135 μM
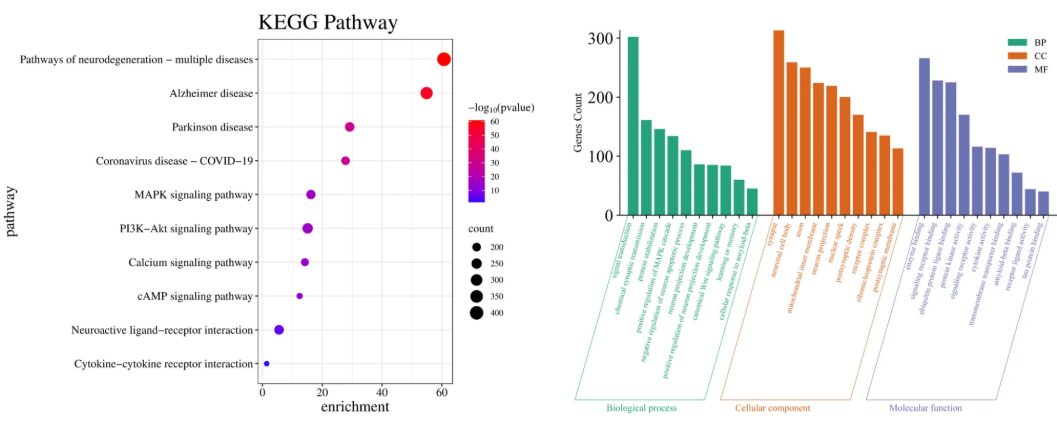
图6. (左)KEGG通路富集分析。富集倍数由横坐标和颜色深度表示,基因富集数量由气泡大小表示。(右)GO富集分析,包含生物过程(绿色)、细胞组分(橙色)和分子功能(紫色)。柱状图长度表示该条目在富集分析中的重要性。
为阐明其作用机制中的关键氨基酸残基,进行了分子对接研究。如图7d所示,化合物1的C-12位羰基通过氢键与Aβ的LYS16残基结合;图7e与7f显示,化合物13的OH-12与残基Pro285形成氢键,化合物14则与Trp82、Trp430、Trp440残基形成三个氢键。结果表明,化合物中羟基或羰基药效团与靶蛋白关键氨基酸残基间的氢键形成,主导了其显著的生物活性。

图7. (a)化合物1的抗Aβ聚集活性(阳性对照:姜黄素,IC50值为1.073 ± 0.125 μM)。(b, c)化合物13(b)和14(c)对BuChE活性的剂量依赖性抑制(阳性对照:多奈哌齐,IC50值为3.71 ± 0.106 μM)。(d)分子对接研究的化合物1与Aβ(PDB代码:1IYT)三维结构及相互作用。(e, f)分子对接研究的化合物13(e)和14(f)与BuChE(PDB代码:4TPK)三维结构及相互作用。
本研究采用分子网络、MS-DIAL及自建数据库联用策略,从橙花瑞香(D. aurantiaca)中系统筛选具有新颖结构及生物活性的倍半萜类成分。在该策略指导下,成功鉴定出15个具多样环系结构的愈创木烷型倍半萜化合物,证明了分子网络、MS-DIAL与自建数据库相结合在快速发现新颖生物活性天然产物方面的巨大潜力。
通过对所有分离化合物进行体外及计算模拟评估,揭示了其在胆碱酯酶抑制及抗Aβ蛋白聚集方面的活性。基于化合物结构新颖性及潜在治疗价值,本研究不仅丰富了倍半萜类化合物的化学多样性,更为开发新型神经系统疾病治疗药物提供了全新化学骨架。

往期回顾
基于CASE-DFT对浅黄枝衣中质子缺失型Chlorodepsidones的结构解析及Flavicansone的结构修订

04-23 Dr. Dai
MSTD系列显微镜专用电动滑台:显微镜下图像分毫必现04-23 光电行业都会关注
展会回顾|“融两业共生之力 筑湾区超级枢纽”2026大湾区创新生态大会04-22 谱临晟
荧飒光学践行企业社会责任,赋能光电人才高质量培养04-22 荧飒光学
天平安装丨现场直击地震对天平的影响,几十台天平瞬间“跳动”?04-22 小普
报名通知丨英斯特朗塑料力学测试高阶培训研讨会04-22 英斯特朗
成都科林分析邀您共赴TFF·2026酒类风味分析与感官评价暨创新技术论坛,期待与您相遇!04-22
吉艾姆4月双展齐发 | 武汉科仪展+脂在浙里研讨会04-22
应用笔记 | 基于Flex自动化平台的多体液胞外囊泡分离及EV蛋白质组学分析流程04-21 肖伟弟 曾嘉明
CCMT2026开展即高能 | Equator-X™ 双模式测量仪引爆全场04-21
世界地球日,查看地球的【愿望清单】04-21
【前沿激荡,智汇北京】IGC 2026圆满落幕,益世科生物共绘细胞基因治疗新蓝图04-21
硬核方案护航核安全|衡昇质谱斩获核材料检测装备大奖04-21
会议预告 | 英盛生物邀您共赴2026第六届北京临床质谱论坛!04-21
Turbiscan在陶瓷3D打印粘结剂分散稳定性表征中的应用及关键意义04-21 大昌华嘉
GranuCharge (粉体静电分析仪)用于研究湿度对粉末表面性能的影响04-21 大昌华嘉
生物打印鼻软骨的理想材料:GelMA水凝胶的力学与细胞外基质平衡新探索04-21 大昌华嘉生命科学
Biolin Theta系列接触角测量 | 如何在表面表征应用中使用接触角:前进角04-21 大昌华嘉
大昌华嘉科学仪器荣获Novasina 2025年度“成长与创新先锋奖”!04-21 大昌华嘉
会议预告|华大吉比爱邀您共赴第六届北京临床质谱论坛04-21 华大吉比爱