Adv Sci项目文章 | 南方科技大学附属第二医院杨亮团队揭示细菌群体感应机制
2023-10-25 10:48:36, 莲莲看 北京青莲百奥生物科技有限公司


巨噬细胞吞噬后绿脓杆菌诱导群体感应机制
为了鉴定绿脓杆菌被宿主免疫细胞吞噬后新合成的蛋白,作者采用pulsed-SILAC蛋白质组学方法,分别用L-lysine-13C6 15N2 (Lys8)和L-lysine (Lys0)标记进入宿主细胞前后的细菌蛋白质组。通过使用这种脉冲标记方法,多达196种蛋白质被确定为细胞内特异性新合成蛋白质。这些蛋白按功能分组,在4小时的感染过程中,一系列毒力相关蛋白在细胞内合成,包括众所周知的群体感应调节因子LasR。
为了验证巨噬细胞吞噬后绿脓杆菌最后群体感应(QS)机制的激活,作者接下来研究了最后群体感应报告系统是否在细胞内感染中确实表达。结果表明,绿脓杆菌在巨噬细胞吞噬后积极表达其群体感应机制,从而可能分泌大量群体感应调节的毒力产物,从内部调节宿主细胞功能。
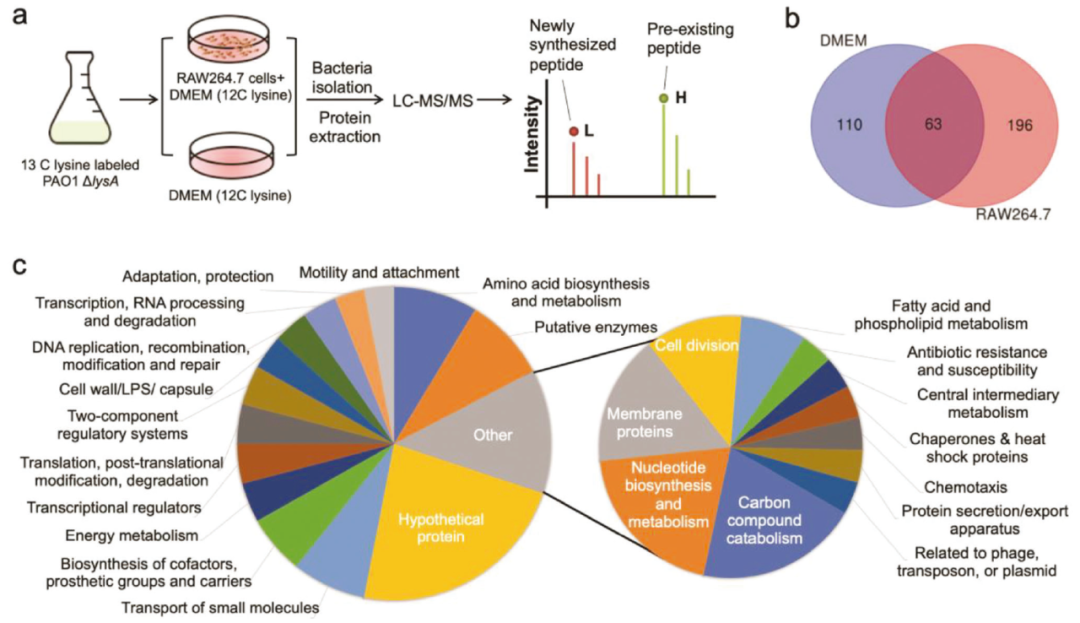
图1 绿脓杆菌在巨噬细胞吞噬后诱导群体感应机制
绿脓杆菌分泌抑制先天免疫信号通过猝灭激活的细胞质先天免疫接头
QS系统控制多种毒力因子的表达,包括鼠李糖脂、凝集素、毒素、蛋白酶,如弹性酶B (LasB)、蛋白酶IV (PIV)和弹性酶a (LasA)。MyD88是先天免疫应答中重要的适配蛋白,在对绿脓杆菌感染的识别和应答中起重要作用。巨噬细胞是一种在检测和消除感染中起核心作用的免疫细胞,在巨噬细胞的背景下,MyD88通过toll样受体(TLRs)参与介导对绿脓杆菌的反应。当绿脓杆菌被TLR4识别后,TLR4受体复合体招募MyD88。MyD88作为一种适配蛋白,促进其他蛋白的募集和激活,包括IRAK(白细胞介素-1受体相关激酶)家族成员在识别绿脓杆菌TLR4受体复合物募集MyD88。IRAK蛋白的激活触发一系列磷酸化事件和蛋白-蛋白相互作用,最终导致NF-κB等转录因子的激活。这些转录因子驱动炎症反应相关基因的表达,如促炎细胞因子和趋化因子。作者进一步用BG-647标记MyD88死亡结构域(DD),用BG-488标记IRAK2-DD种子,通过检查IRAK2的缓慢迁移,推断出哪个MyD88DD是活跃的。通过对绿脓杆菌和其他11种不同细菌的分泌物的筛选,发现绿脓杆菌的分泌抑制了MyD88 DD和含有CSRD的细胞凋亡相关斑点样蛋白(ASC)的寡聚。进一步的实验表明,信号抑制是通过切割免疫适配器的死亡结构域。
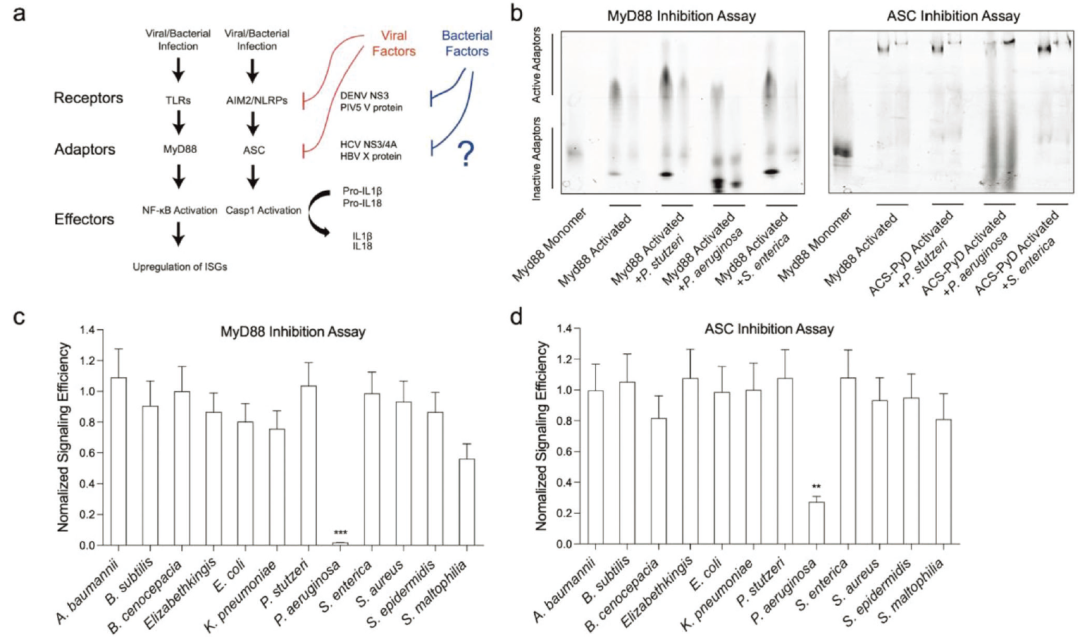
图2 绿脓杆菌分泌抑制先天免疫信号通过淬灭激活的细胞质先天免疫适配器
绿脓杆菌分泌的先天免疫抑制因子的生化特性
绿脓杆菌分泌物混合物与MyD88 DD复合物孵育,发现缓冲条件的改变对先天免疫抑制活性有显著影响。SDS-PAGE分析显示,这些条件破坏了实验中观察到的裂解活性,表明抑制剂是离子依赖性的,对极端pH环境敏感。此外,MyD88 DD的裂解具有时间依赖性,可以在反应的较早时间点通过热处理进行淬灭。通过SEC和离子交换,成功分离出两个有助于MyD88 DD降解活性的组分。通过胰蛋白酶辅助质谱分析,确认该酶的身份是LasB,这是绿脓杆菌的一种著名的由群体感应调节的毒力产物。
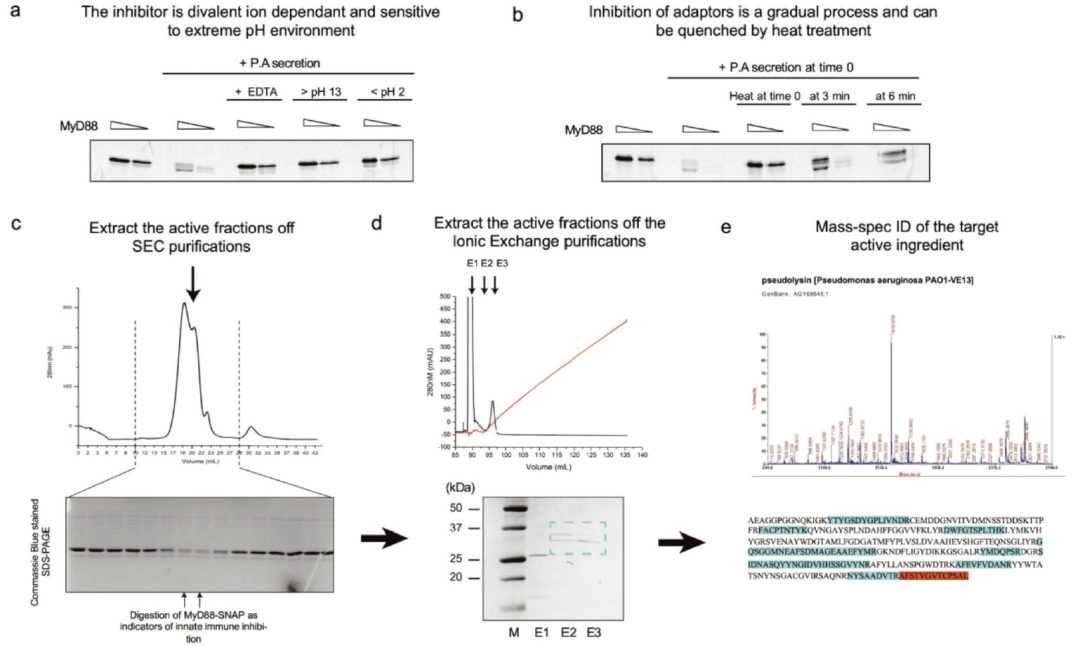
图3 绿脓杆菌分泌的先天免疫抑制因子的生化特性及纯化
单LasB对绿脓杆菌分泌的先天免疫抑制活性起作用
lasB缺陷菌株(ΔlasB)的分泌物不能破坏MyD88信号在体外的传播,而lasB的补充恢复了表型。MyD88 DD过表达的HEK293T细胞通过NF-κB荧光素酶报告基因检测进行评估显示,40 nM的LasB足以使MyD88 dd诱导的炎症反应减少一半。胰蛋白酶控制表明LasB的蛋白水解活性对先天免疫蛋白具有特异性。如果将LasB与血清成分预先孵育或热处理,这种免疫抑制活性就消失了,这表明有必要将折叠良好的LasB直接送入宿主细胞以抑制先天免疫反应。此外,阴性染色电镜(EM)实验证实纯化的预活化MyD88低聚物被LasB消化成无活性片段。因此,LasB直接消化先天免疫适配器,从而抑制免疫反应。此外,与其他先天免疫适配蛋白相比,MyD88形成相对较小和灵活的结构,这可能使其更容易受到蛋白酶的抑制。
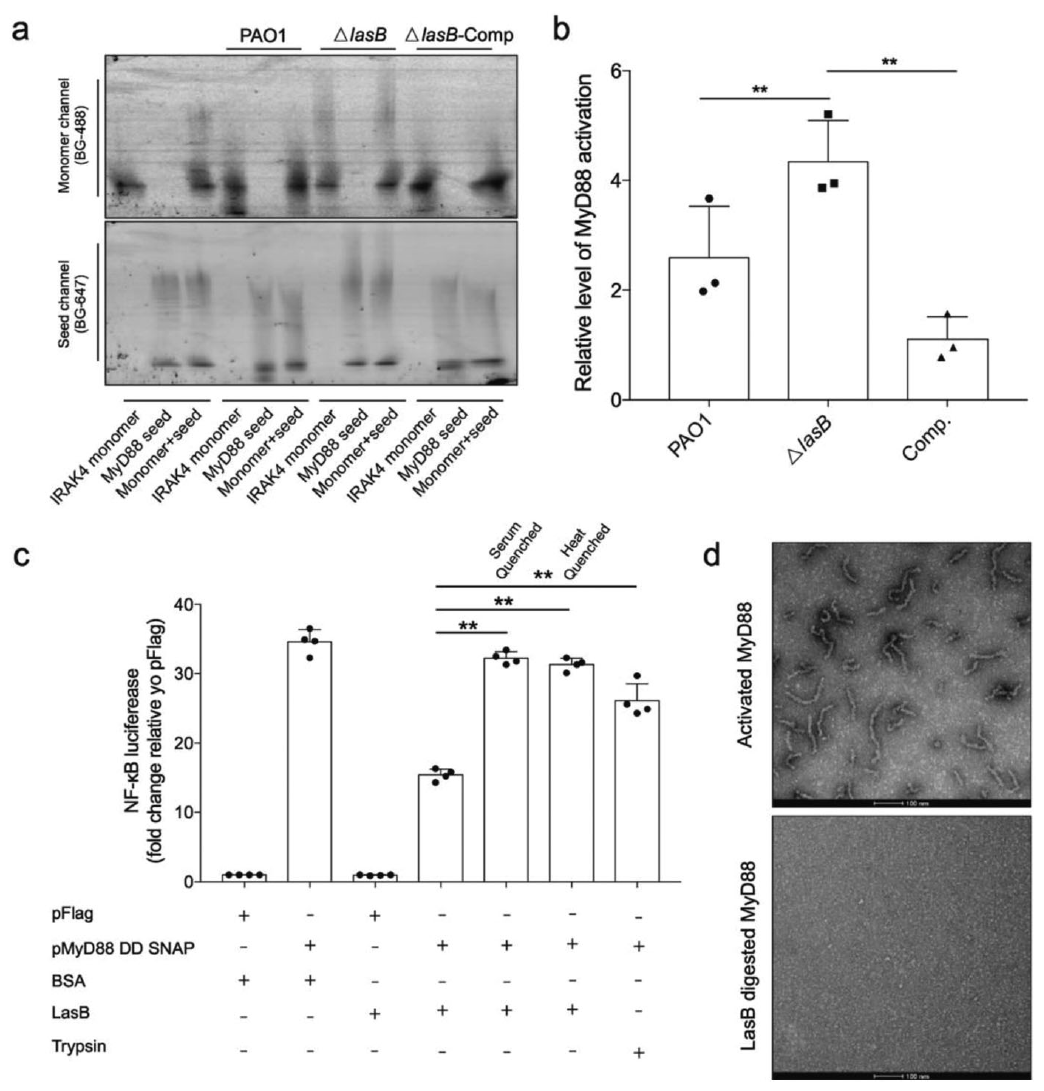
图4 LasB单独负责绿脓杆菌分泌的先天免疫抑制活性
LasB抗炎特性的结构论证
对几种M4家族肽酶底物结合袋周围的氨基酸序列比对显示出高度的序列相似性。这使得很难确定LasB能够降解宿主细胞中的死亡域接头的原因。利用以往积累的结构数据,作者比较了这些LasB同源物的活性位点袋的三维结构。LasB在所比较的所有同源物中具有最宽的底物结合槽,颈距为6.9 Å。更大的开口将赋予这种锌金属蛋白酶适应α螺旋的刚性环或转折点的能力,这是死亡结构域中潜在的切割位点。在LasB催化袋附近引入了轻微的点突变,略微缩小了底物结合槽的颈部距离。所有三个突变体(H223Y、A113S和V137L)都将沟槽缩小了0.6-0.8 Å,这种扰动足以显著破坏LasB消化MyD88 DD的酶活性。
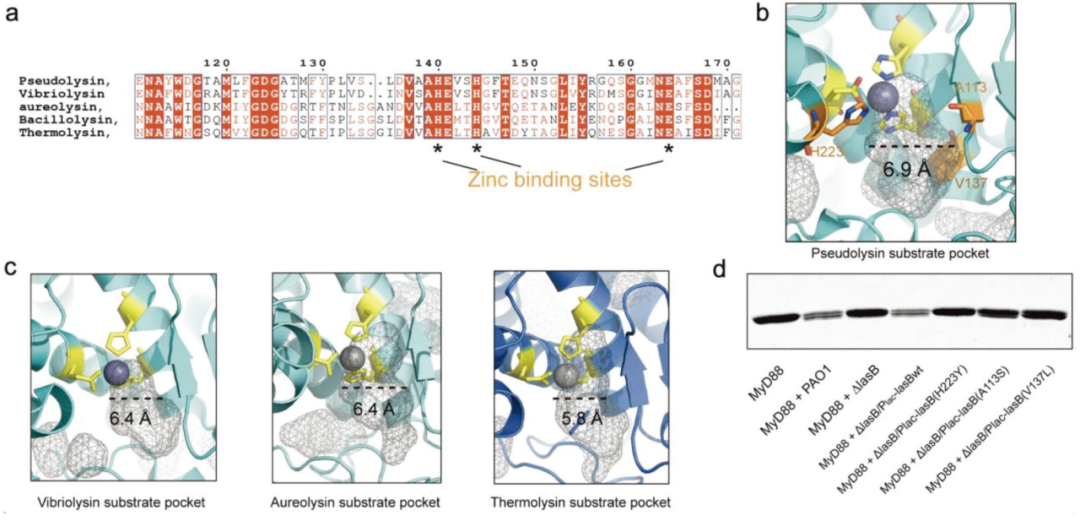
图5 LasB抗炎特性的结构论证
MyD88 DD在A24L25位点被LasB切割,这是稳定活性信号低聚物界面所必需的
采用质谱分析方法鉴定LasB酶切后的肽片段,发现A24L25也符合LasB底物偏好的一般预测,开始成为最可能的裂解位点。该基序位于MyD88 DD的第一个α-helix的扭结处,使其更容易被外部因素访问。同时,这个潜在暴露的基序对于稳定多聚体MyD88复合物非常重要,这对于TLR信号传导至关重要。在重组实验中,与野生型MyD88-DD相比,MyD88 DD (A24S)明显抵抗LasB消化。平行控制突变体(G106D)不赋予MyD88 DD这种保护。在细胞荧光素酶实验中,MyD88野生型和A24S突变体在HEK239T细胞中过表达,当纯化的≈40-50 nM LasB电穿孔到细胞中时,MyD88 (A24S)比野生型构建体更能抵抗免疫抑制。
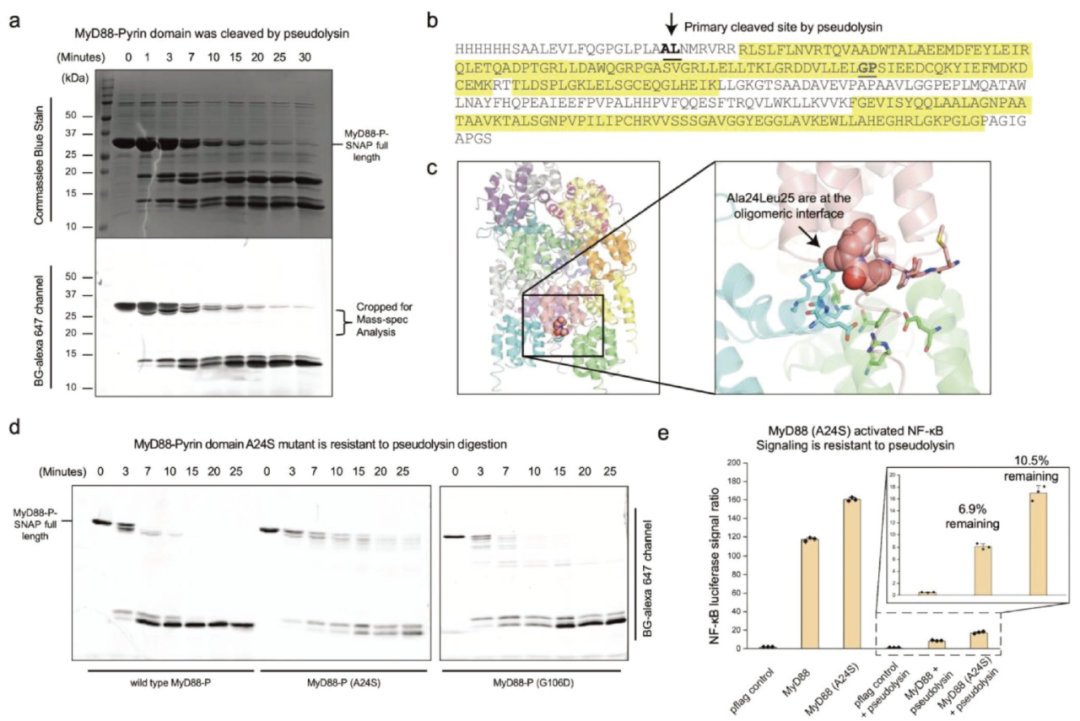
图6 MyD88 DD在A24L25位点被LasB切割
LasB在绿脓杆菌胞内感染过程中表达和分泌
抗flag免疫印迹显示LasB在宿主细胞质内积极分泌。腺苷酸环化酶(Cya)报告基因实验进一步证实了LasB的分泌Cya2-400报告结构域被融合到全长LasB蛋白的C端。将编码LasB融合蛋白和Cya2-400结构域的基因插入到phd20t质粒中,编码这些蛋白的基因由LasB启动子驱动转录。感染产生LasB-Cya融合的绿脓杆菌导致巨噬细胞内RAW 264.7 cAMP水平急剧增加。相比之下,感染单独产生Cya的绿脓杆菌不影响cAMP水平。
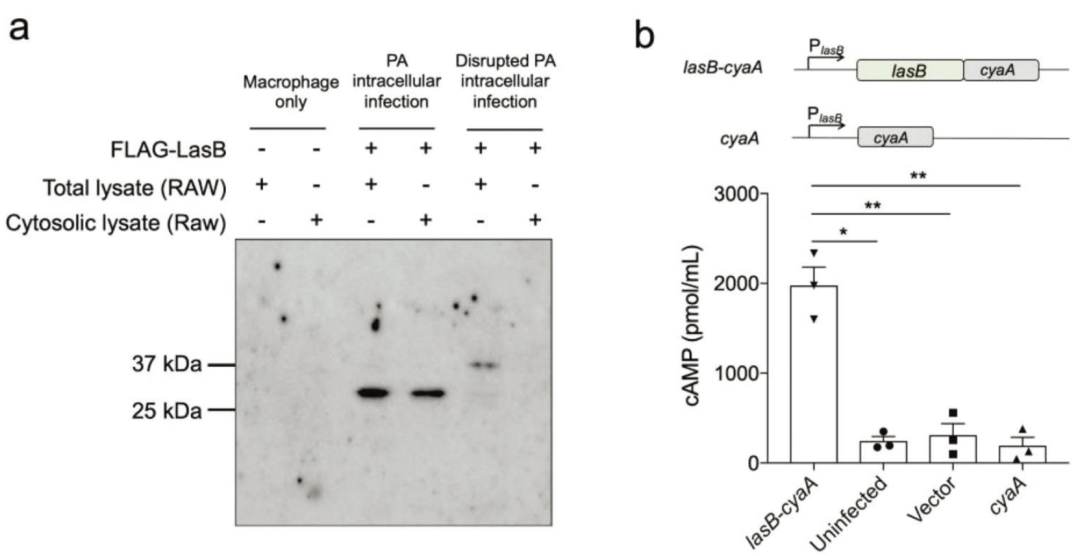
图7 在绿脓杆菌的机会性细胞内感染过程中,LasB被分泌并活跃
LasB降解巨噬细胞MyD88并降低TNF-α水平
分别用PAO1、ΔLasB和LasB补体菌株感染RAW264.7细胞,并通过western-blotting分析细胞内MyD88的水平。与PAO1和补体菌株相比,感染ΔlasB菌株后细胞内MyD88水平升高。MyD88靠近TLR复合体,下游信号也被检查。Western blot数据显示,与ΔlasB突变体相比,PAO1减弱了NF-κB的p65亚基的磷酸化,而对ERK、JNK、p38的磷酸化没有影响。进一步测定了TNF-α的产生,发现与PAO1和补体菌株相比,ΔlasB菌株的TNF-α水平显著升高。
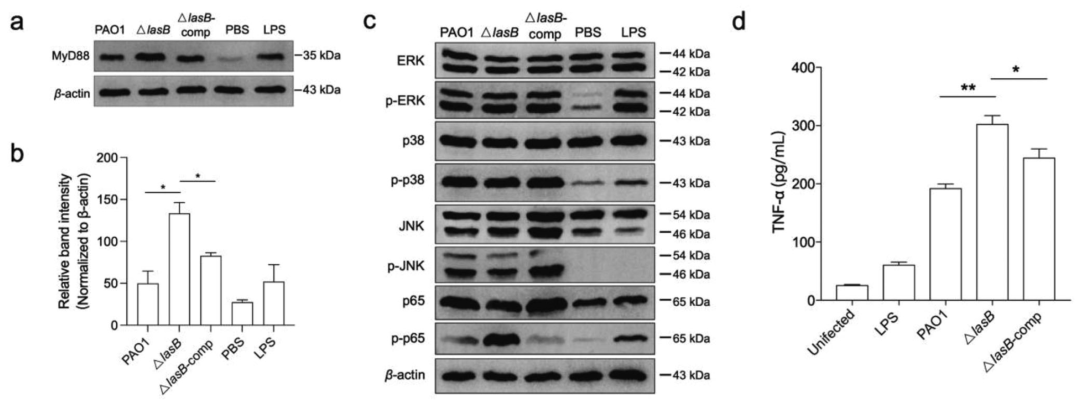
图8 LasB降解巨噬细胞中的MyD88,降低TNF- α水平
研究结论
在这项研究中,作者观察到绿脓杆菌能够在吞噬巨噬细胞后存活,并通过代谢标记蛋白质组学方法诱导群体感应机制。然后采用重组方法分离出能够在受体-适配器阶段淬灭宿主免疫信号的细菌因子。在筛选细菌分泌组后,发现绿脓杆菌群体感应调节产物LasB可以消化和阻止多种先天免疫受体的寡聚化,从而在它们的机会性细胞内感染期间沉默免疫反应。该研究认为LasB和其他相关的细菌蛋白酶在慢性细胞感染的建立中发挥了重要作用。此外,该研究还揭示了细菌蛋白酶如何降解和去除宿主细胞中的死亡结构域蛋白寡聚物,从而帮助细菌逃避先天免疫反应的机制。
【参考文献】
Duan X, Boo ZZ, Chua SL et al. A Bacterial Quorum Sensing Regulated Protease Inhibits Host Immune Responses by Cleaving Death Domains of Innate Immune Adaptors. Adv Sci (Weinh). 2023 Oct 23:e2304891.
关于青莲百奥







04-11 NanoTemper
FAQ系列1_Ligand梯度稀释04-11 NanoTemper
直播预告 | PR Panta助力ADC研发04-11 NanoTemper
FAQ系列2_结合buffer04-11 NanoTemper
这份证书,让你脱颖而出 | 福立仪器2024年NTC培训计划启动,赶快报名抢占先机!04-10
Peak微课堂 | 氢气发生器应用合集04-10 毕克气体
探索“哈希中源纪”,精美好礼随心兑!04-10
QbD1200+总有机碳分析仪在第三方医疗器械清洁度检测中的应用04-10
博赛德VOCs检测技术与ODS等新污染物检测前沿探究 · 全国巡回技术交流会之南京站即将拉开帷幕04-10 市场部
距离医疗新质生产力亮相CMEF还有一天!04-10
上海仪电·科学仪器诚邀您参加第61届中国高等教育博览会04-09
GE医疗中国携手国家传染病医学中心及上海感染与免疫科技创新中心搭建肝病全周期防控路径04-09
BDO一网打尽 | 过程监测篇04-09 福立仪器
赛默飞与您相约第三届中国国际氢能及燃料电池高峰论坛暨展览会04-09
赋能创“芯” | 赛默飞电子气体气相色谱分析解决方案04-09 飞飞
【线上培训】沃特世GPC/APC聚合物分析技术应用交流会04-09 沃特世技术服务
Empower控制质谱使用小贴士(四)| 培训讲座进行时04-09 沃特世技术服务
纤微影示,涵蕴起初 — 急诊篇 二04-09 岛津医疗
Nature Reviews:如何降低mRNA-LNP的毒性风险(3):开发非临床毒理研究模型04-09
Nature Reviews:如何降低mRNA-LNP的毒性风险(1):消除IVT RNA的免疫原性04-09