Givosiran的非临床药代动力学及吸收、分布、代谢和排泄,第一个被批准的N-乙酰半乳糖胺偶联RNA干扰治疗药物
2023-06-20 21:58:32 艾杰尔-飞诺美(Agela & Phenomenex)

Givosiran是一种N-乙酰半乳糖胺偶联的RNA干扰治疗药物,靶向肝脏中的5''-氨基乙酰酸合成酶1mRNA,目前已上市用于治疗急性肝性卟啉症。本文对Givosiran的非临床药代动力学、吸收、分布、代谢和排泄特性进行了表征。Givosiran皮下给药后完全吸收,血浆消除半衰期相对较短(t1/2;少于4小时)。血浆暴露与剂量近似成比例增加,重复给药后无积累。在所有被测试的种属中,血浆蛋白结合率呈浓度依赖性,人体临床相关浓度约为90%。Givosiran主要通过去唾液酸糖蛋白受体介导的摄取分布到肝脏,并且在肝脏中的t1/2明显更长(~1周)。Givosiran通过核酸酶而非细胞色素P450 (P450)同工酶进行跨种属代谢,无人类特有代谢物。Givosiran代谢形成一个主要的活性代谢物,从反义链的3''端丢失一个核苷酸,AS(N-1)3''Givosiran,与Givosiran具有同等效力。肾脏排泄和粪便排泄是消除Givosiran的次要途径,在大鼠和猴子的排泄物中分别有大约10%和16%的剂量被完整地回收。Givosiran不是P450同工酶的底物、抑制剂或诱导剂,也不是摄取和大多数外排转运蛋白的底物或抑制剂。因此,Givosiran介导P450同工酶和药物转运体的药物-药物相互作用的潜力较低。
体内实验
所有的动物实验都是按照当地、州和联邦法规进行的,并得到了Alnylam制药公司动物护理和使用委员会的批准。对雄性和雌性大鼠、食蟹猴进行Givosiran单次静脉注射或单次和多次皮下注射给药,剂量水平在每项研究中规定。开始给药时,大鼠约7~12周龄,体重160~325g。猴为2至8岁,体重为2至6公斤。大鼠和猴静脉注射剂量为10mg/kg,大鼠皮下注射剂量为1~10mg/kg,猴皮下注射剂量为1~30mg/kg。收集血浆、尿液、乳汁、粪便和其他组织(肝脏、肾脏等)样本,冷冻保存在约-70°C以待分析。
代谢物研究与样品制备
通过液相色谱-高分辨质谱法(LC-HRMS)对Givosiran的代谢物进行分析,并对Givosiran原药及其主要代谢物AS(N-1)3''Givosiran进行定量分析,AS(N-1)3''Givosiran是一种双链代谢物,由反义链3''端失去一个核苷酸形成,分析方法同之前工作。简单地说,根据制造商推荐的方案,使用Clarity OTX 96孔板(Phenomenex, Torrance, CA)对血浆、尿液、乳汁、粪便匀浆和组织匀浆进行固相萃取处理,提取的样品通过LC-HRMS进行分析。
结果
Givosiran大鼠血浆药代动力学
图1 大鼠单次皮下给药(10mg/kg)后,Givosiran(A)和AS(N-1)3′Givosiran(B)的个体和平均(±sd)血浆浓度-时间曲线。Givosiran和AS(N-1)3′Givosiran血浆Cmax值分别为1.06和0.190μg/ml。Givosiran和AS(N-1)3′Givosiran血浆AUClast值分别为3.00和0.626h·μg/ml。AUClast评估的AS(N-1)3’Givosiran的血浆暴露约为Givosiran暴露的21%。在达到Cmax后,Givosiran和AS(N-1)3’Givosiran浓度分别以t1/2值3.0和8.2h下降。注:误差条表示sd定量下限=10ng/ml。低于定量浓度下限的按0处理。结果显示为单个动物(开放符号)和平均值(封闭圆圈;N=3)。
Givosiran猴血浆药代动力学

图2 单次皮下注射Givosiran(30mg/kg)后,雄性猴Givosiran(A)和AS(N-1)3''Givosiran(B)的个体和平均(±sd)血浆浓度-时间曲线。血浆中Givosiran和AS(N-1)3′Givosiran的Cmax值分别为2.42和1.67μg/ml。Givosiran和AS(N-1)3′Givosiran血浆AUClast值分别为26.4和19.4h·μg/ml。AUClast评估的AS(N-1)3’Givosiran的血浆暴露量约为Givosiran暴露量的74%。在达到Cmax后,Givosiran和AS(N-1)3’Givosiran浓度分别在5.5和5.1h的t1/2值下降。注:误差条表示sd定量下限=10ng/ml。结果显示为单个动物(开放符号)和平均值(封闭圆圈);N=3)。
药物在大鼠的体内分布
图3 静脉给药和皮下给药(10mg/kg)后大鼠的平均肝脏浓度-时间分布。单次静脉或皮下给药后,Givosiran的肝脏AUClast值分别为5390小时·µg/g和12600小时·µg/g。单次皮下给药10mg/kg后肝脏暴露量显著高于静脉给药,表明皮下给药后肝脏吸收更有效。IV,静脉注射;SC,皮下。误差条表示每个时间点每组4只动物。
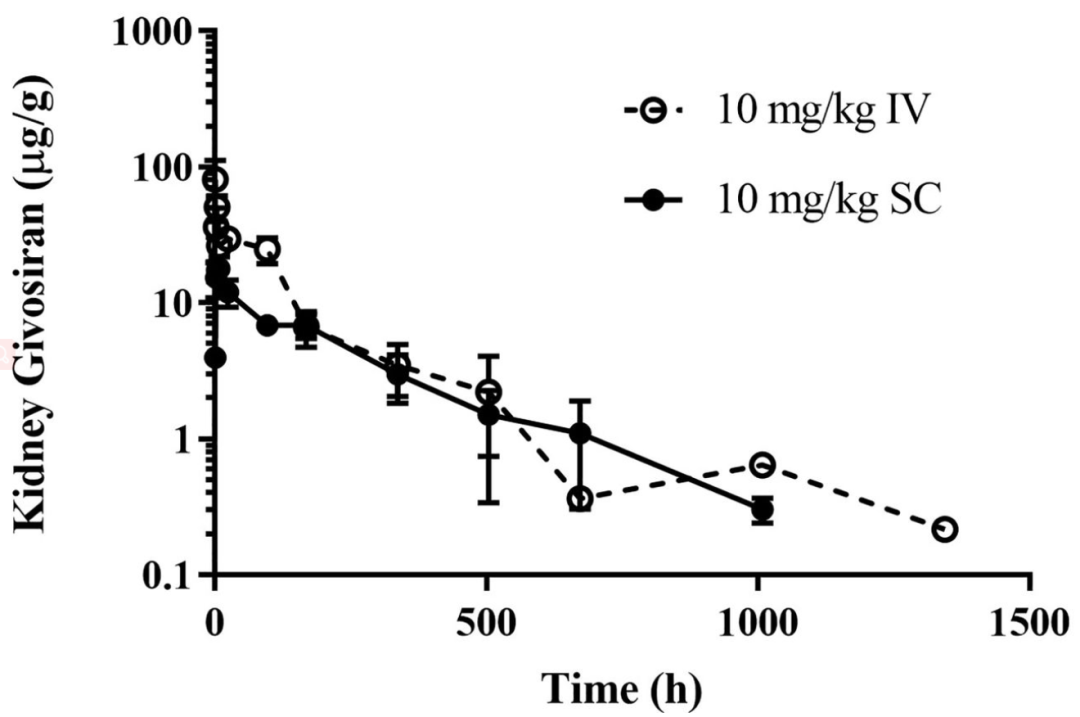
图4 静脉给药和皮下给药(10mg/kg)后大鼠平均肾浓度与时间曲线的关系。单次静脉或皮下给药后,Givosiran的肾AUClast值分别为5440小时·µg/g和3190小时·µg/g。静脉给药后较高的血浆浓度导致肾脏中较高的Givosiran浓度,Givosiran从血浆中的分布可能是被动扩散(即没有asgpr介导的摄取)。IV,静脉注射;SC,皮下。误差条表示每个时间点每组4只动物。
药物在猴的体内分布
图5 静脉给药(10mg/kg)和皮下给药(1-10mg/kg)后,猴的平均肝脏浓度-时间分布。单次皮下给药(1、5或10mg/kg),在给药后672-1008小时的肝脏中可检测到Givosiran,在给药后8至24小时观察到最大肝脏浓度。单次静脉或皮下给药10mg/kg后,肝脏AUClast分别为4220小时·µg/g和28500小时·µg/g。单次皮下给药后肝脏AUClast比相同剂量静脉给药后高约7倍,表明皮下给药后肝脏摄取比静脉给药更有效。IV,静脉注射;SC,皮下。每个时间点每组N=2只动物。
结论
综上所述,在体外和体内对Givosiran的PK和ADME特性进行了表征。Givosiran在非临床种属的体内试验,以及人类和动物基质的体外试验中,显示出相似的PK和ADME特性模式。汇总数据表明,皮下给药Givosiran导致siRNA充分暴露于预期的靶器官(肝脏)。总的来说,PK和ADME研究为毒理学研究的解释提供了支持,帮助描述了每月一次2.5mg/kg给药方案下人体对Givosiran的处置特征,并支持临床使用Givosiran治疗急性肝性卟啉症。
参考文献
Drug Metabolism and Disposition July 2021, 49 (7) 572-580;
07-01 英斯特朗
连载 | 药物一致性评价与粒度分析(三)07-01 欧美克仪器
【仪器百科】LS-909丨干湿二合一激光粒度分析仪07-01 欧美克仪器
标准物质解决方案 | PFASs(全氟及多氟化合物)06-29
第九期阿尔塔有约 | 环境专题【新污染物:PFAS】技术研讨会精彩回顾及提问解答06-29
“绿色技术范式”,分析化学未来发展方向——访中国分析测试协会副理事长、辽宁省分析科学研究院原院长刘成雁教授06-29 转载仪器信息网
华西医院-标准型数显脑立体定位仪、双通道体温维持仪、体式显微镜安装完成06-29 迈越生物
科鉴检测助力2家仪器企业获得首批产品可靠性认证证书06-28 科鉴检测
德国耶拿:锂电池生命周期分析解决方案06-28 德国耶拿
AI已来!生命科学本科教学如何紧跟技术浪潮06-28 Opentrons
盛瀚售后,五星级服务的秘诀是什么?06-28 SHINE
专为汽车制造商打造的柔性解决方案——实现制程控制06-28
西北工业大学-脑立体定位仪安装完成06-28 迈越生物
会议邀请 | 第九届海上检验医师论坛06-28
卓立要闻 | 创新发展ing…6月卓立“大事小情”速览06-28 光电行业都会关注
打造信任合作伙伴!2024年度卓立汉光客户满意度调查开启06-28 光电行业都会关注
如何挑选适用于三阶光学非线性的测量系统?Z扫描测量系统来助力!06-28 光电行业都会关注
招聘启事—中国科学院沈阳自动化研究所微纳光学测量表征技术课题组06-28 光电行业都会关注
谱育科技作为主要完成方 荣获2023年度国家科学技术进步一等奖和二等奖06-28 点击关注→
仪器原理丨顶空仪与吹扫捕集仪科普小知识06-28 天美色谱