机器学习与生物信息--XGBoost
2022-09-14 07:59:50 上海吉凯基因医学科技股份有限公司
随着遗传学、育种学的不断发展和人类基因组计划和分子生物学的日益发展,生物学数据在短短的几十年里得到了爆发式地增长。比如生物信息学里面的:回归分析、随机森林、支持向量机等算法,都是比较成熟的应用了。在最近阅读文献得过程中,小编发现了一篇材料学科的文献,文中用到的XGBoost算法与小编之前钻研过的两篇生信文章的算法十分相似,那么今天就给大家剖析一下当生物信息遇见机器学习,会有什么火花,自己文章中又如何加入这些有趣的机器学习方法,增加创新,帮助你投不出去纯分析文章增加成本。

一、生物信息学数据
研究的数据类型,可以分为基因型数据(GenoType Data)、表达量数据等;其中基因型数据则是通过WGS、WES、基因芯片数据获得的。
如今,基因组信息被广泛用于癌症的精确治疗。由于个体类型的组学数据只代表单一观点,存在数据噪声和偏差,因此需要多种类型的组学数据来准确预测癌症预后。然而,由于多组学数据中存在大量冗余变量,但样本量相对较小,有效整合多组学数据具有一定的挑战性。
二、机器学习与生物信息学数据的结合
我们越来越多地看到机器学习在生信文章中应用,例如针对数据寻找可用的模式然后进行预测。通常,这些预测模型用于操作流程以优化决策过程,但同时它们也可以提供关键的洞察力和信息来报告战略决策。
机器学习的基本前提是算法训练,提供特定的输入数据时预测某一概率区间内的输出值。请记住机器学习的技巧是归纳而非推断——与概率相关,并非最终结论。
构建这些算法的过程被称之为预测建模。一旦掌握了这一模型,有时就可以直接对原始数据进行分析,并在新数据中应用该模型以预测某些重要的信息。模型的输出可以是分类、可能的结果、隐藏的关系、属性或者估计值。
如果我们关心的是估算值或者连续值,预测也可以用数字表示。输出类型决定了最佳的学习方法,并会影响我们用于判断模型质量的尺度。
谁对机器学习方法进行监督?机器学习方法可以是有人监督也或者是无人干预的。区别不在于算法是否可以为所欲为,而是是否要从具备真实结果的训练数据中学习——预先确定并添加到数据集中以提供监管——或者尝试发现给定数据集中的任何自然形态。大多数企业使用预测模型,对训练数据使用监督方式,而且通常旨在预测给定实例——邮件、人员、公司或者交易是否属于某个有趣的分类——垃圾邮件、潜在买家、信用良好或者获得后续报价。
如果在开始之前你不是很清楚在寻找什么,那么无人干预的机器学习方法能够提供全新的洞察力。无人干预的学习还能够生成集群与层次结构图,显示数据的内在联系,还能够发现哪些数据字段看起来是独立的,哪些是规则描述、总结或者概括。反过来,这些洞察能够为构建更好的预测方法提供帮助。
构建机器学习模型是一项反复练习的过程,需要清理数据和动手实验。目前市场上正在涌现一些自动和有向导的模型工具,它们承诺降低对数据科学家的依赖性,同时在常见领域获得最高的投资回报率。然而这里面真正的差别很可能需要你自己去发现。
随着深度学习技术的发展,自动编码器被用于整合多组学数据,提取具有代表性的特征。然而,由于数据噪声的影响,生成的模型很脆弱。此外,以往的研究通常集中在单个癌症类型,而没有对泛癌症进行全面的测试。
三、算法介绍
1.GBDT(Gradient Boosting Decision Tree):在数据分析和预测中的效果很好。它是一种基于决策树的集成算法。
2.Boosting:Boosting指把多个弱学习器相加,产生一个新的强学习器。经典的例子有:adaboost, GBDT, xgboost等。如果每一个弱学习器用 来表示的话,那么Boosting的强学习器就可以表示为:
通俗的来说就相当于把多个学习器串联(bagging是并联)。接下来,我们就介绍一下xgboost算法。
3. XGBoost:
XGBoost本质上是一个GBDT,但是力争把速度和效率发挥到极致,所以叫X (Extreme) GBoosted
XGBoost 树定义:
举例
预测一家人对口红的喜爱程度,考虑到年龄相比,年轻人更可能喜欢口红,男性和女性相比,女性更喜欢口红,故先根据年龄大小区分成年人和未成年人,然后再通过性别区分开是男是女,逐一给各人在口红喜好程度上打分,如下图所示。

XGBoost的核心算法思想不难,基本就是:
1. 不断地添加树,不断地进行特征分裂来生长一棵树,每次添加一个树,其实是学习一个新函数f(x),去拟合上次预测的残差;
2. 当我们训练完成得到k棵树,我们要预测一个样本的分数,其实就是根据这个样本的特征,在每棵树中会落到对应的一个叶子节点,每个叶子节点就对应一个分数;
3. 最后只需要将每棵树对应的分数加起来就是该样本的预测值。
通俗来讲,XGBoost是GBDT算法的一种改进,是一种常用的有监督集成学习算法;是一种伸缩性强、便捷的可并行构建模型的GradientBoosting算法。
原理是:在GBDT目标函数的基础上加入惩罚项,通过限制树模型的叶子节点的个数和叶子节点的值来降低模型复杂度,从而防止过拟合,二分之一是为了求导方便。t是树的棵数,obj为损失函数。

(看不懂没关系,理解这样做的目的就好~大体步骤是为了防止过拟合,二阶泰勒展开公式计算,给出了新的树的划分标准,用的是损失函数的增量)
目的:找到第t颗树是如何搭建的所以我们的期望是损失函数只和第t颗树有关系。
XGBoost出现较早,但在交叉学科,比如生物、化学、材料等领域的应用较少,抓住这个新颖的方向可能为你的论文增添看点,XGBoost支持开发语言:Python、R、Java、Scala、C++等。
XGBoost 的最佳信息来源是该项目的官方 GitHub 库:https://github.com/dmlc/xgboost
四、文献与总结
第一篇
Improving protein-protein interactions prediction accuracy using XGBoost feature selection and stacked ensemble classifier

期刊:《Computers in Biology and Medicine》
影响因子及中科院分区:IF:3.434,中科院三区
发表日期:2020年7月
作者单位:青岛科技大学
1.算法方法
(1)作者提出了一种新的预测蛋白质-蛋白质相互作用的方法——StackPPI
(2)融合PAAC、AD、AAC-PSSM、Bi-PSSM和CTD提取物理化学、进化和序列信息
(3)采用XGBoost特征选择方法消除冗余,保留最优特征子集
(4)首次利用RF、ET和LR构建了堆叠集成分类器。
2.数据
训练集:
幽门螺杆菌( Helicobacter pylori),正负样本各位1458
酵母菌(Saccharomyces cerevisiae),正负样本各位5594
测试集:
人相互作用对数量为1412
老鼠相互作用对数量为313
秀丽隐杆线虫相互作用对数量为4013
大肠杆菌相互作用对数量为6954
基因评估数据集:
Wnt相关通路:96个作用对
疾病特异性:108个作用对
3.结果解读:
流程图:
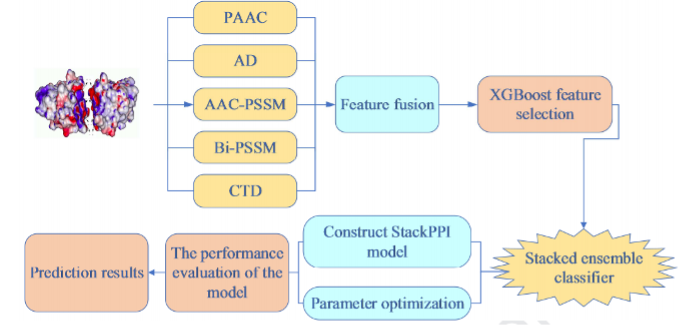
(1)确定参数m

首先根据图2 (A)可以看出,随着参数λ值的变化,两个数据集的ACC值是不同的。当λ =11时,StackPPI s 精准性(ACC)在幽门螺旋杆菌和酿酒酵母菌数据集上达到一个全局最大值,而在幽门螺旋杆菌数据集上λ =9时达到最大值。利用平均精度得到StackPPI (λ =11)中PAAC的最优参数λ。图2 (B)绘制了不同m下Moreau-Broto、Moran和Geary自相关描述子ACC的变化。m = 8时,幽门螺旋杆菌ACC最高,m = 9时,啤酒酵母StackPPI最高。通过平均预测精度,在StackPPI中设m = 9, 自相关描述符AD(Moreau-Broto、Moran和Geary、氨基酸组成位置特异性评分矩阵)的维数为21*9=189。
(2)数据降维方法的评估与选择
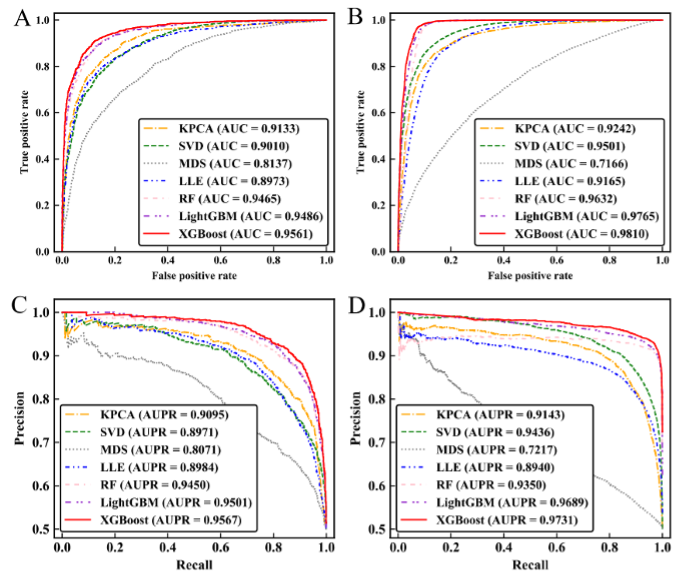
对于不同的数据集,采用不同的方法,并通过受试者工作曲线进行有效性的评估。为了选择最优分类算法,将堆叠集成分类器与逻辑回归(LR)、k -最近邻(KNN)、AdaBoost、随机森林(RF)、支持向量机(SVM)和XGBoost进行比较。其中KNN方法的邻域设为5,SVM采用径向基核,AdaBoost、RF和XGBoost的''n_estimators''分别为500、500和500。所以在本节中,使用XGBoost作为分类器。为了进一步验证StackPPI,作者对不同的分类器进行统计检验。报告了LR、KNN、AdaBoost、RF、SVM、XGBoost与堆叠集成分类器相比在ACC、MCC和AUC指标上的p值。

4.总结:蛋白质-蛋白质相互作用(Protein-protein interaction, PPIs)在蛋白质组水平上参与了大多数细胞活动,这篇文章作者使用机器学习的算法与生物信息学数据结合探讨和预测蛋白相互作用。作者提出了一个名为StackPPI的预测框架。首先使用伪氨基酸组成、自相关描述符(Moreau-Broto、Moran和Geary、氨基酸组成位置特异性评分矩阵), Bi-gram位置特异性评分矩阵以及组成、转移和分布对生物相关特征进行编码。其次,采用XGBoost算法去除特征噪声,并通过梯度提升和平均增益进行降维;最后,通过StackPPI(一种由随机森林、极度随机树和逻辑回归算法组成的堆叠集成分类器开发的PPIs预测器)对优化后的特征进行分析。
第二篇
Integrating multi-omics data through deep learning for accurate cancer prognosis prediction

期刊:《Computers in Biology and Medicine》
影响因子及中科院分区:IF:3.434,中科院三区
发表日期:2021年5月
作者单位:中山大学
1.算法方法:
(1)DCAP方法的体系结构:将多组癌症数据的高维特征输入DAE网络,得到具有代表性的特征,然后利用这些特征通过Cox模型估计患者的风险。考虑到临床难以获得多组数据,进一步利用mRNA数据构建XGboost模型来拟合估计的风险。构建的模型用于预测独立数据集中的癌症患者风险。此外,基于XGboost和差异表达分析鉴定出的基因,我们鉴定出9个与乳腺癌预后高度相关的预后标志物。
(2)自动编码器
(3)XGBoost特征制作风险模型、
2.数据:
(1)TCGA癌症数据
(2)GEO癌症数据
3.结果解读:
流程图:

(1)使用数据类型与方法的比较与筛选
如表2所示,DCAP在10倍CV和独立检验中获得了基本相同的c指数值,对15种癌症的平均值分别为0.678和0.665。结果表明,该方法具有较好的鲁棒性。我们进一步详细介绍了每种组学类型在DCAP中的贡献。
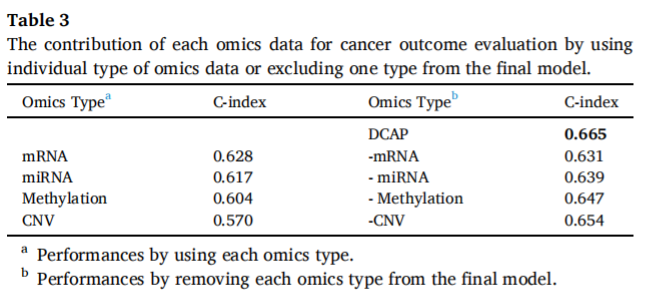
如表3所示,单类组学数据中,mRNA表现最好,平均c指数为0.628,CNV表现最差,c指数为0.570。miRNA和甲基化分别排在第2位和第3位。一致认为,当从DCAP中排除一种组型时,mRNA引起的c指数值下降最大,从0.665下降到0.631,而排除CNV引起的c指数下降最小。这些结果表明,mRNA在鉴别高危患者中起着最重要的作用,而CNV的作用最小。平均而言,使用多组学的预后预测比仅使用mRNA数据的预后预测提高了5.9%。
(2)案例研究
作为一个案例研究,研究人员将作者的方法应用于包含最多样本的乳腺癌(BRCA)。为了验证DCAP-XGB构建的乳腺癌预后预测模型,在GEO数据库中收集的三个外部乳腺癌数据集GSE2990、GSE9195和GSE17705上对模型进行了测试。

如图3A所示,3个数据集预测的高风险和低风险组与生存曲线明显分离,p值均在0.05以下,c指数相近(0.602、0.605、0.611)。这些结果表明了作者轻加权风险预测模型的稳健性。
根据DCAP对高危和低危人群的划分,我们鉴定出159个DEGs,其中有45个风险基因下调,114个风险基因上调(图3B)。159个DEGs中,有57个(35.9%)基因经过了文献证实与乳腺癌相关。
用XGboost模型选择的223个基因作图,发现9个DEGs重叠,其中7个(77.8%)基因(ADIPOQ、NPY1R、CCL19、MS4A1、CCR7、CALML5和AKR1B10)与乳腺癌相关(表5)。对于剩下的2个基因(ULBP2和BLK),虽然没有文献直接证明与乳腺癌预后相关,据报道,ULBP2的诱导与p53的药理学激活触发抗癌先天免疫反应[27]有关,而BLK是一个真正的能诱导肿瘤的原癌基因,适合于BLK驱动淋巴瘤的研究和体内[28]中新型BLK抑制剂的筛选。
4.总结:
如今,基因组信息被广泛用于癌症的精确治疗。由于个体类型的组学数据只代表单一观点,存在数据噪声和偏差,因此需要多种类型的组学数据来准确预测癌症预后。然而,由于多组学数据中存在大量冗余变量,但样本量相对较小,有效整合多组学数据具有一定的挑战性。随着深度学习技术的发展,自动编码器被用于整合多组学数据,提取具有代表性的特征。然而,由于数据噪声的影响,生成的模型很脆弱。此外,以往的研究通常集中在单个癌症类型,而没有对泛癌症进行全面的测试。在这里,作者使用去噪自编码器来获得多组数据的鲁棒表示,然后使用学习到的代表性特征来估计患者的风险。应用美国癌症基因组图谱(TCGA)中的15个癌症样本,结果表明该方法比传统方法平均提高6.5%。考虑到实际操作中难以获得多组数据,作者进一步通过训练XGboost模型,仅使用mRNA数据拟合估计的风险,发现模型平均c -指数为0.627。以乳腺癌预后预测模型为例,分别在基因表达综合数据库(Gene Expression Omnibus, GEO)的3个数据集上进行独立检验,结果显示该模型能够显著区分高危患者和低危患者。根据作者的方法划分的风险亚组,识别出9个与乳腺癌高度相关的预后标志物,其中7个基因已被文献综述证实。从而得出结论,本研究构建了一个准确、稳健的多组学数据综合预测肿瘤预后的框架。此外,它也是发现癌症预后相关基因的有效途径。
第三篇
XGBoost model for electrocaloric temperature change prediction in ceramics

期刊:中国科学院上海硅酸盐研究所主办的《npj Computational Materials》
影响因子及中科院分区:IF:12.3,中科院一区
发表日期:2022年7月
作者单位:卡耐基梅隆大学
1.算法方法:
(1)XGBoost算法
2.数据:
(1)电热材料(EC)数据集:EC材料主要有三大类:聚合物、陶瓷和聚合物陶瓷复合材料。作者建立了EC陶瓷的数据集,因为它们的成分种类繁多。作者从现有文献中提取信息,因为大多数材料成分没有出现在知名的材料数据库。该数据集包含45篇论文中的97篇材料,可以在GitHub48上以csv格式访问。数据集的快照以及数据收集和模型构建步骤的流程图如图所示。
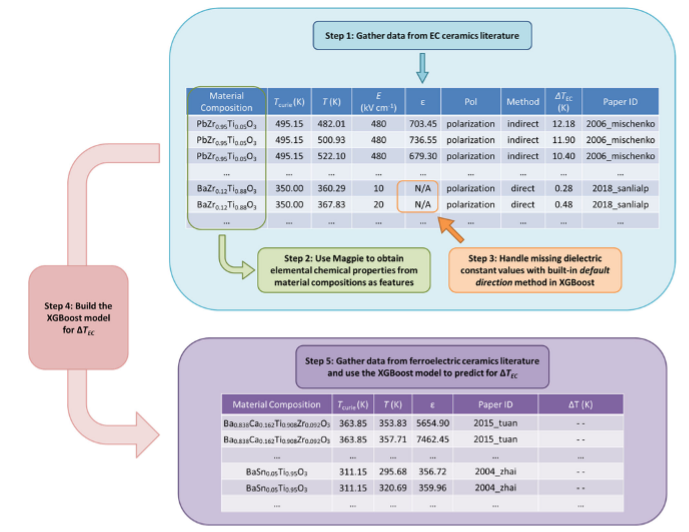
3.结果解读:
(1)数据预处理
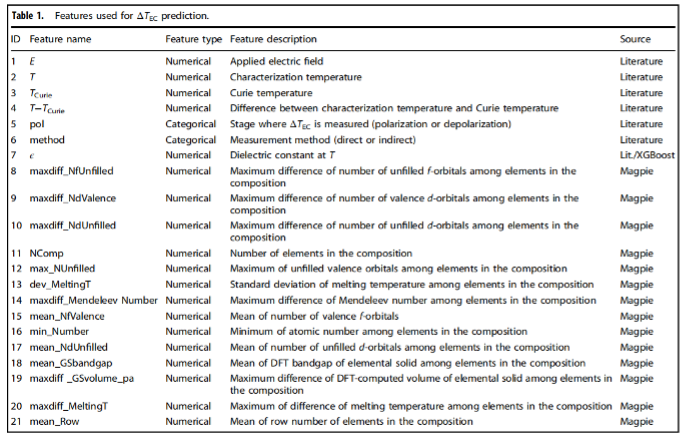
经过一些预处理步骤去除了不合格的数据,研究人员有4406个数据点,每个数据点包含表1所列的21个特征(7个实验条件/材料性能特征和14个混杂特征)。要预测的标签是在给定条件下(即T和E)的ΔTEC。

在图2a中,收集的数据被绘制为全尺度表征温度的函数。在图2b中,ΔTEC在0 2k范围内的数据点被绘制成T-TCurie的函数。不同的颜色代表不同的材料组成,标记尺寸与所施加的电场成比例。这些EC材料的温度变化相对较小,中值为0.36 K,平均值为1.07 K。97个材料中有3个最大,远远超过了第二大最大值13 K。这三种材料被标记为异常值,在构建模型时被排除,除非另有规定。
(2)XGBoost造模
用于ΔTEC预测的XGBoost回归模型(详见方法XGBoost回归部分)是通过6912种组合的网格搜索建立的最佳超参数集(表2)。由于XGBoost无法进行外推,只能对之前在训练历史中遇到的情况做出合理的预测,除非特别说明,否则ΔTEC值最低和最高的材料将被迫出现在训练集中。作者构建了三个模型,并根据其随机种子进行区分。虽然,正如预期的那样,XGBoost模型预测PbZr0.97La0.02(Zr0.95Ti0.05)O3不能比训练集的最大值高于ΔTEC,但它们对PbZr0.95Ti0.05O3的预测能力范围都高于ΔTEC。这一观察结果表明,XGBoost模型从基础物理中学习,可以作为定性预测和改进新材料搜索的有用工具。
(3)验证模型
根据94个EC陶瓷在特征空间中的距离,将其分为训练数据集和测试数据集。首先将EC材料的复杂特征投影到二维t分布随机邻域嵌入(t-SNE)空间上。然后对94种材料的投影进行k-means聚类。通过将聚类内平方和作图为k的函数,并将“肘部”标识为k,通过肘部法确定最佳k值为3。为所有数据分配一个聚类标签。从每个聚类中选取75%的材料作为训练数据,其余25%作为测试数据。
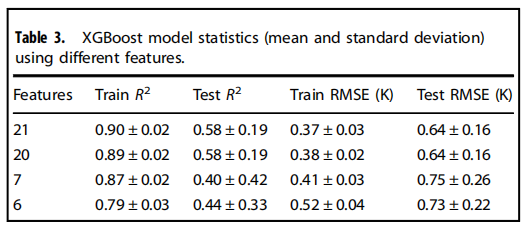
不同数量的特性用于构建模型。分别为表1中的21个特征、去掉介电常数的20个特征,去掉所有混杂特征的7个特征,去掉介电常数和所有混杂特征。对于每个特性集,使用相同的超参数,但不同的随机种子和训练/测试分割训练了100个XGBoost模型。R2和RMSE结果(平均值和标准差)汇总在表3中,其中每一行的数据对应100个模型。
(4)模型对于杂质的特征分析
我们对XGBoost对 ΔTEC模型进行了特征分析,其奇偶图如图3所示。
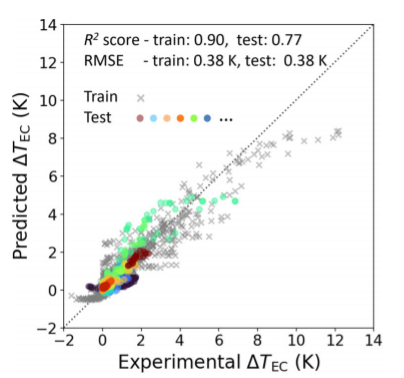
基于杂质的特征重要性由XGBoost通过测量在使用特征的所有分割处的总增益(即精度的提高)来计算。特征重要性值越高,说明特征越重要。
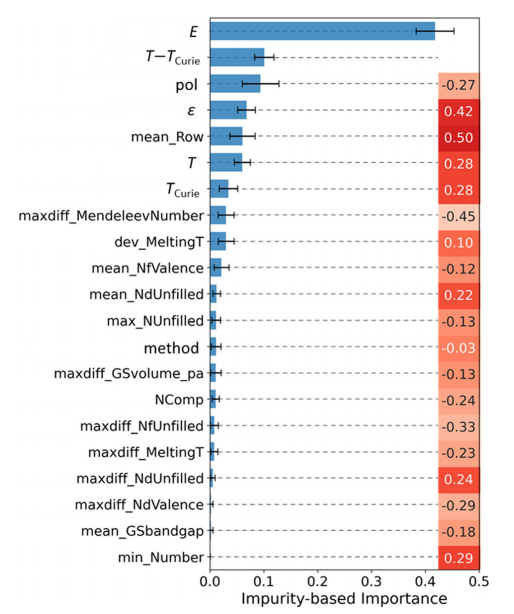
基于杂质的特征重要性如图4所示。应用电场E排名第一,其次是T-Tcurie居里。这些观察结果与已知的物理学一致。16个混杂功能协同帮助模型区分不同的材料。因此,直接解释单个特征可能是困难的。XGBoost模型在所有145个混杂特征(或110个非零方差特征)中选取了这四组特征。作者的XGBoost模型是严格规范化的(即,在节点的拆分中使用了树修剪和限制),该模型对包含不相关的特性并不敏感。通过使用XGBoost ΔTEC模型进行预测,其奇偶图如图3所示。
4.总结:
XGBoost梯度提升通过梯度下降算法将预测误差降至最低,并产生一组弱预测模型(决策树)形式的模型。在训练过程中,梯度提升每次增加一个新的回归树,以减少残差(即模型预测与标签值之间的差值)。模型中现有的树保持不变,这降低了过拟合的速度。作者在本研究中建立了一个极端梯度Boosting (XGBoost)机器学习模型,根据陶瓷的成分(由混杂元素属性编码)、介电常数、居里温度和表征条件,预测陶瓷的电热(EC)温度变化。
根据实验文献,建立了97个EC陶瓷的数据集。通过对特征空间中的聚类数据进行采样,模型对测试数据的决定系数为0.77,均方根误差为0.38 K。特征分析表明,该模型捕捉了有效电导率材料的已知物理特性。混杂特征有助于模型区分材料,元素电负性和离子电荷被确定为关键特征。将该模型应用于66种电导率未得到表征的铁电体。确定了在室温和100kv /cm下EC温度变化大于2 K的无铅候选材料。
五、文章小结
基因组学是一门跨学科的生物学学科,它可以量化生物体的所有基因,并研究它们对生物体的相互作用影响,如今机器学习已广泛应用于基因组学研究,使用已知的训练集来预测数据类型的结果,同时深度学习和深度学习模型可以预测并且降维分析能力更强更灵活,在适当的训练数据下,深度学习可以在人工干预较少的情况下自动学习特征和规律。深度学习目前也成功应用调控基因组学、突变检测和致病性评分,可以提高基因组数据的可解释性,并将基因组数据转化为可操作的临床信息、改进疾病诊断方案、了解谁应该使用什么药物和药物,从而最大限度地减少药物副作用,最大限度地提高疗效,由于涉及的变量太多,人工的统计分析较慢,而深度学习可以帮助缩短过程。
而XGBoost训练速度极快,内存友好,可以计算每个特征的重要性,这对于特征筛选、模型可解释性、模型透明、模型调优等都有好处;XGBoost还可以以明文的形式保存树模型,方便模型可视化和调优,这么多的优点,赶紧趁它没有广泛应用在生物信息学领域的时候放到你的论文中吧!
(注:文章转自生信人微信公众号)


1.实验技术干货
2.蛋白质组学研究
3.腺病毒简介及应用
4.临床基础研究思路解析
5.组织特异性腺相关病毒
6.单细胞测序
7.慢病毒实验操作指南
8.悬浮细胞专用病毒
9.靶点设计/数据库教程
10.测序技术研究与应用
11.非编码RNA研究技术与应用
12.腺相关病毒选择/应用
13.表观遗传研究
14.文章解析
15.国自然课题设计思路解析
16.生物信息分析及工具
17.外泌体研究
18.肿瘤免疫研究
19.高分文章
20.吉凯病毒神经方向应用案例
06-18 TESCAN中国
解决临床创新“卡脖子”难题,丹纳赫重磅推出医院创新转化解决方案!06-18
徕卡精准空间生物学解决方案 第三弹06-18 郑晓业、童昕
618 嗨购一起“徕” | 网上商城活动来袭!06-18 徕卡显微系统
【培训活动】显微镜成像高阶培训系列(一) 共聚焦多维度成像技术解决方案06-18 徕卡显微系统
【展会】第十五届中国医师协会骨科医师年会06-18 徕卡显微系统
【直播预告】第十届电子显微学网络会议06-18 徕卡显微系统
【案例研究】双视野光片显微镜,适用于类器官及胚胎06-18 徕卡显微系统
议程确定|第四届锂离子电池热测试主题研讨会06-18
文献解读|安徽理工大学马衍坤教授团队《煤炭学报》:震动载荷多次作用下烟煤孔裂隙结构演化特征试验研究06-18 纽迈分析
中国的新质生产力正在服务全球市场!GE医疗北京基地CT交付量达3.5万台/套06-17
兰格ACHEMA 2024展会回顾:探寻精密流体传输市场新机遇06-17
宝英科技推出VOCs治理设施精细化管理实施方案,助力重点行业企业绿色升级06-17 宝英科技
赛默飞X先锋“肽”势:多肽药物研发与创新研讨会06-17
一省检验检测行业营收突破160亿元!06-17
仪器租赁 | 沃特世 液质联用,月租金52000元起06-17
不同行业实验室“以旧换新”,涉及哪些仪器设备?06-17
目标!检验机构总营收超100亿!06-16
一张“A4纸”售价高达万元,这家第三方检测公司是怎么做到的?06-15
GLMY创想仪器丨参加东北地区铸造年会06-15 国产精密仪器厂家