转录组专题 | 转录组结题报告解读+文章撰写描述
2022-07-15 07:40:51, 小维 武汉迈维代谢生物科技股份有限公司

转录组作为研究基因表达的利器,已经成为实验室的常用工具。那么拿到转录组的数据之后,动辄就有几十页的分析报告,几十项分析内容。每个图表的含义是什么,哪一些分析内容是需要重点关注的呢?真正可以用到文章里的分析内容又有哪些?
本次我们对转录组结题报告里的比较重点的分析内容进行介绍,以方便各位老师快速入手转录组结题报告。
一.转录组结题报告整体介绍

00. Report,网页版的结题报告,用来整体了解项目的信息及快速查找需要的图表。
01. Genome_annotation,基因组的fasta文件、基因组的注释信息、基因在各数据库中的注释结果, 基因组的cDNA序列,CDS序列,氨基酸序列。
02. Quality_control,测序数据量统计及数据质控结果。
03. Alignment,与参考基因组的比对统计。
04. Novel transcript,新基因预测,在染色体上的位置、各数据库注释结果、序列。
05. Quantitation,基因的原始readcount、FPKM、及在所有数据库中注释结果汇总,样本的相关性分析,PCA分析,小提琴图,表达量密度分布图。
06. Different_analysis,比较组合差异分析结果,富集分析结果,kmeans结果,韦恩图结果。
07. Alternative_splicing,可变剪切分析,类型及各类型数量、所在基因及基因在各数据库中的注释结果。
08. Variant_analysis,变异位点分析包括SNP和Indel。
09. WGCNA,WGCNA分析结果。
10. PPI,蛋白互作网络分析。
转录组数据可重点查看Quantitation,Different_analysis,WGCNA三个文件,如果对基因结构比较感兴趣,可查看Variant_analysis变异位点,Alternative_splicing可变剪切文件。
二.Quantitation解读
结果内容包括:所有样本的相关性分析,所有样本的PCA分析,所有样本的基因表达量及数据库注释信息,所有样本FPKM表达量密度图和表达量分布图。

2.1 all_cor所有样本的相关性分析
用来评价组内生物学重复样本的重复性,还可以评估差异表达基因的可靠性和辅助异常样品的筛查。将皮尔逊相关系数r(Pearson’s Correlation Coefficient)作为生物学重复相关性的评估指标。R绝对值越接近1,说明两个重复样品相关性越强。

图1.样本相关性图
2.2 gene_pca所有样本的主成分分析
主成分分析(Principal Component Analysis,PCA)是一种无监督模式识别的多维数据统计分析方法,用来判断组内样本的重复性和组间样本的差异。

图2.样本的PCA图
2.3 gene_expression.annot所有样本比对上的所有基因及注释信息
包括基因ID,基因的FPKM值,基因的counts值,以及在不同数据库的注释信息,这个表是所有后续差异基因分析的原始结果。

三.Differential_analysis部分解读
结果内容主要包括:
all DEG:所有差异基因及注释列表和所有差异基因的聚类热图;
*VS*:每个差异分组中的差异基因列表,差异基因MA图,差异基因火山图,差异基因GO注释及富集,差异基因KEGG注释及富集,差异基因KOG注释,差异基因聚类热图,差异分组的GESA富集分析;
基因的K-means分析:包括基因的kmeans图片,及所有基因分簇类别和注释结果;
Venn:韦恩图及每个韦恩图对应到的差异分组的基因表达量和注释结果。

3.1 差异基因筛选
差异基因的筛选标准默认为FDR<0.05,FC≥2或≤0.5。
如果项目内某个差异分组差异基因的个数过多,可以联系公司将筛选参数设置的更严格。如果觉得差异基因个数过少,原因可能是:① 比较组之间本身差异就比较小;② 由于组内生物学重复差导致组间差异变小。所以出现差异基因少的时候,可以先查看组内生物学重复的情况,如果是原因①,那可以视研究需要决定要不要降低筛选标准,如果是原因②,那就要考虑是不是要剔除样本。
注:差异基因应该有多少个并没有标准,也并不是越多越好,只要能解释研究的问题就可以。
3.2 差异基因火山图
火山图展示差异分组里检测到的基因总数以及显著上调和下调差异基因个数。火山图横坐标表示基因表达倍数变化,纵坐标表示基因的显著性水平。红色的点代表上调的差异基因,绿色的点代表下调的差异基因,蓝色的点代表非差异表达的基因。

图3.差异基因火山图
3.3 差异基因的聚类热图
聚类热图展示差异基因及在样本间的聚类。横坐标表示样品信息及层次聚类结果,纵坐标表示差异基因及层次聚类结果。红色表示高表达,绿色表示低表达。多组比较的情况下,两两之间的差异个数没法直观展示。建议筛选完最终关注的基因时,再通过热图展示。

图4.差异基因的聚类热图
3.4 差异基因韦恩图
韦恩图可以展示不同差异分组之间的共有和特有差异基因的个数。韦恩图非重叠区代表该差异分组特有的差异基因,重叠区代表重叠的几个差异分组共有的差异基因。

图5.三个差异分组的三元韦恩图
韦恩图相关文献案例
We compared the differentially expressed genes (DEGs) in pink tea petals among different floral development stages. In total, 23,868 DEGs were identified, which were differentially expressed at different tea flower development stages. The number of differentially expressed genes had very high variance among different development stages. The largest number of DEGs was found betweenBTP1 and BTP4, where 9563 DEGs were identified. The libraries of BTP1 vs. BTP2, BTP1 vs. BTP3,BTP1 vs. BTP4, and BTP1 vs BTP5 respectively had 946, 4272, 9563 and 9087 DEGs. Among those DEGs, the four libraries had 611 common DEGs. There were 513 upregulated DEGs in BTP1 vs. BTP2, 1763 upregulated DEGs in BTP1 vs. BTP3, 4257 upregulated DEGs in BTP1vs. BTP4, and 4062 upregulated DEGs in BTP1vs. BTP5.(Ying Hong et al.BMC Plant Biol., 2019.)

图6.文章里展示的韦恩图分析
3.5 差异基因的GO注释及GO富集分析
Gene Ontology(简称GO)是基因功能国际标准分类体系。作为基因本体联合会(Gene Onotology Consortium)所建立的数据库,它旨在建立一个适用于各种物种的,对基因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准。GO分为分子功能(Molecular Function)、生物过程(biological process)、和细胞组成(cellular component)三个部分。横坐标为GO二级分类,纵坐标为基因的表达量。

图7.差异基因二级条目分类图
一般情况下GO注释的差异基因往往比较多,很难找到与自己研究相关的GO条目。那么可以利用GO富集分析,辅助筛选掉一些富集不显著的条目。GO富集分析的原理为超几何分布,GO-Term显著性富集分析以GO数据库中GO-Term为单位,应用超几何检验,找出与整个基因组背景相比,在差异表达基因中显著性富集的GO-Term。横坐标表示注释到该条目的基因与有注释的基因总数的比例,纵坐标表示GO条目的名称。图形右侧的标签代表GO条目所属的分类。

图8.差异基因GO富集柱形图
GO注释部分相关文献案例
A total of 79,208 (73.5%) unigenes were assigned into 9,945 Gene Ontology (GO) terms and, of these, 67,003, 68,347 and 67,160 were assigned at least one GO term in the ‘biological process’, ‘cellular component’ and ‘molecular function’ categories, respectively. The ‘biological process’ includes ‘cellular protein modification process’, ‘response to stress’, ‘carbohydrate metabolic process’, ‘lipid metabolic process’, ‘secondary metabolic process’, among others. The molecular functions were related to hydrolase activity, catalytic activity, transferase activity, kinase activity, lipid binding. Additionally, we identified the putative biochemical pathways represented in the assembled mango transcriptome. Our analysis showed that a total of 7,740 unigenes were annotated in 461 pathways, including triacylglycerol biosynthesis, phenylpropanoid biosynthesis.(Tafolla-Arellano Julio C et al.Sci Rep, 2017)

图9. 文章中展示的GO注释结果
3.6 差异基因的KEGG注释及富集分析
Kyoto Encyclopedia of Genes and Genomes(KEGG,https://www.genome.jp/kegg)是整合了基因组、生物学通路、疾病、药物、化学物质等信息的综合性数据库。KEGG 将基因组信息和高层次的功能信息有机地结合起来,为基因组测序和其他高通量实验技术产生的大数据提供系统化的分析。通过kegg注释,可以查看差异基因注释到了kegg的哪个通路,以及在通路上的上下调关系。Kegg通路中,红色框标记的酶与上调基因有关,绿色框标记的酶与下调基因有关。蓝色框标记的酶与上调和下调基因均有关。

图10.KEGG通路图
与GO相同,每个差异分组中也会注释到很多条kegg通路,这时候就需要进行kegg通路富集分析,筛选与研究目的比较相关的kegg通路。纵坐标表示 KEGG 通路。横坐标表示 Rich factor。Rich factor 越大,富集的程度越大。点越大,通路富集的差异基因的数量越多。点的颜色越红,代表富集越显著。

图11.Kegg富集气泡图
kegg注释部分相关文献案例:
KEGG pathway enrichment analysis (Q-value <0.05),revealed that these DEGs were mainly enriched in several metabolic processes including phenylpropanoid biosynthesis, lipid metabolism, and amino acid metabolism.(Meng Jie et al.J. Agric. Food Chem., 2019)

图12.文献中展示的Kegg富集分析图
3.7 K-means趋势分析
将相同表达趋势一致的基因归为一类,方便按照大类去筛选关注基因。
如果有表型参数,可以根据表型参数的变化趋势,筛选与表型参数变化趋势一致的,或者趋势相反的簇来重点关注。如果做项目前通过参考文献和前期实验获得了一些关注基因,可以重点关注在同一表达簇中的其他基因(与关注基因富集在相同功能和通路上的其他基因,以及在聚类分析中表达模式跟关注基因相近的其他基因可以一起研究)。
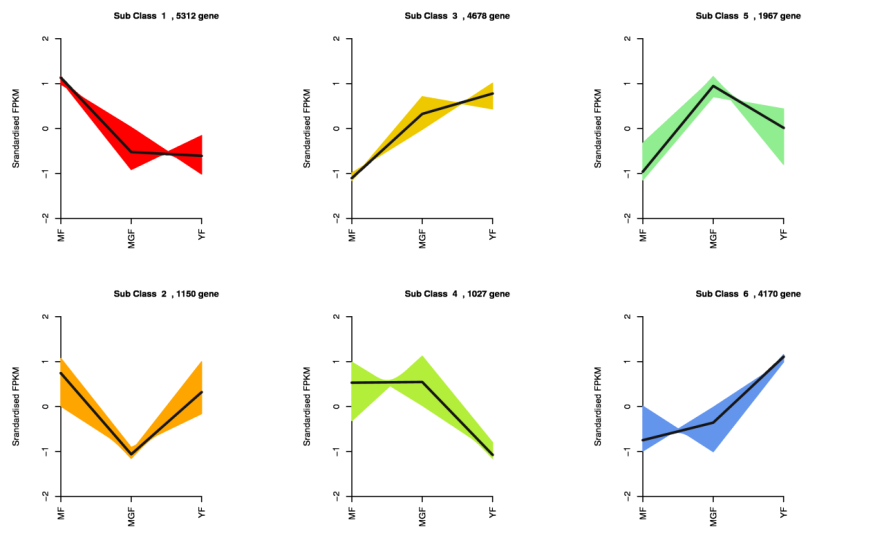
图13.基因K-means分析图
快速入手转录组的结题报告可直接查看重点讲解部分,对于数据细节问题可直接联系所在区域销售。
登录迈维云,查看完整版转录组结题报告
(https://cloud.metware.cn/#/user/login?redirect=%2F)
精彩合集,欢迎收藏
迈维云平台系列课操作教程,公号回复“迈维云”即可

我就知道你“在看”

04-24 汉尧
四方光电荣膺工信部“制造业单项冠军企业”04-24 四方光电
文末惊喜丨你的合成反应“翻过车”吗?这些痛点,中了几个?04-24 奥豪斯
传奇续写:奥豪斯旗下涡旋振荡器Vortex-Genie 2焕新登场04-24 奥豪斯
采用先进防静电技术,最大限度减少干扰04-24 奥豪斯
询价有礼 | 奥豪斯电化学产品解决方案04-24 奥豪斯
官宣!5月16日,第五届量子科仪节暨量子精密测量产业应用峰会与您相约合肥!04-23 CIQTEK
Angew速递:台式easyXAFS原位解析高效析氧反应的定向非晶到非晶重构04-23 Dr. Dai
安捷伦与西北农林科技大学共建“旱区农产品感官科学联合实验室”04-23
五一水机关不关?测测你是水机“杀手”还是“达人”04-23 默克MilliQ纯水
Adv Sci(IF 14.1) | 上海交通大学王慧/李晓光/李辰团队通过时空蛋白质组学图谱揭示食管鳞状细胞癌早期进展的预警信号04-23
在山野间畅快呼吸,于同心处共启新程04-23 毕克气体
MSTD系列显微镜专用电动滑台:显微镜下图像分毫必现04-23 光电行业都会关注
分光光度计怎么用?一步步教你正确操作与数据读取方法04-23 管理员
分光光度计的工作原理详解:从朗伯-比尔定律到现代检测技术04-23 管理员
2026国产化电镜技术前沿与产学研用协同发展论坛顺利召开!04-22 CIQTEK
2026 磁共振技术应用与创新学术交流会圆满落幕!聚焦磁共振前沿,共探国产仪器创新之路04-22 CIQTEK
展会回顾|“融两业共生之力 筑湾区超级枢纽”2026大湾区创新生态大会04-22 谱临晟
安捷伦的地球日|以科学之力,将可持续落实到每一天04-22 安捷伦
荧飒光学践行企业社会责任,赋能光电人才高质量培养04-22 荧飒光学