必看!99% 科研人会忽略的测序致命伤
2020-05-10 07:59:33 安捷伦科技(中国)有限公司

HiSeq X Ten 和 NovaSeq 大大提升了测序通量,但样本标签错配问题着实让人头疼。FFPE、液态活检样本的趋动变异频率实在太低,而建库环节、PCR 环节、测序本身的错误率就和变异频率差不多,如何将它们区分开?
优化的建库方案是解锁各项难题的一把钥匙,解题之前让我们先熟悉一下很多人傻傻分不清,但与上述问题直接相关的两种标签(index)——样本标签和分子标签。

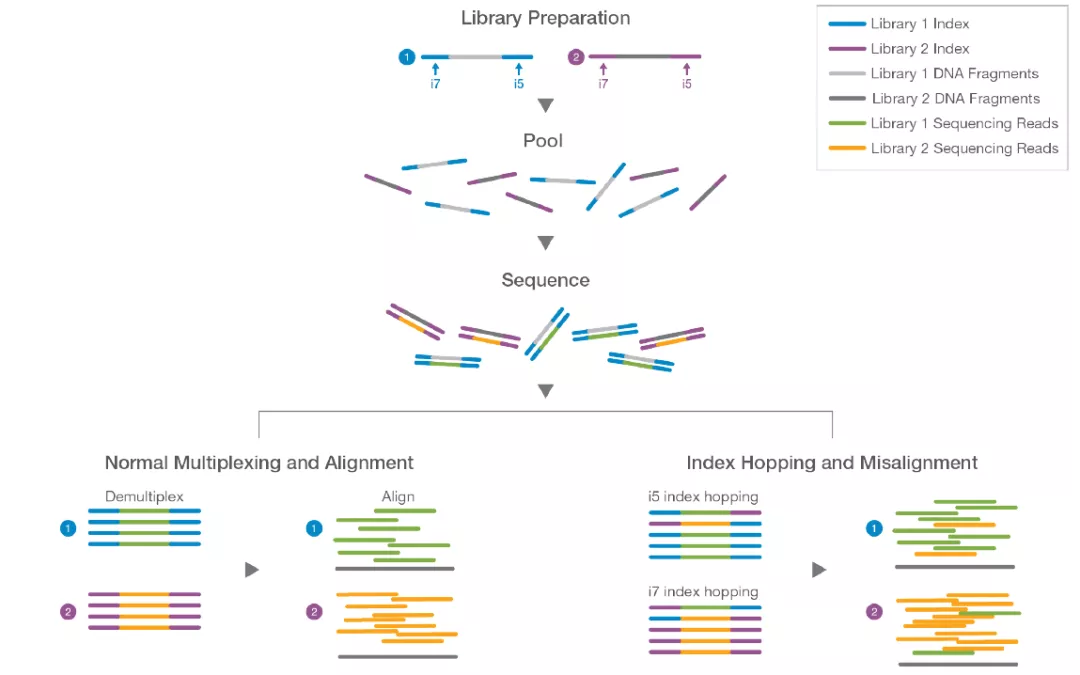
目前最普遍采用的双样本标签,相当于给样本标记加上双保险。然而组合型的双端标签(Combinational Dual Index–CDI)仍然存在标签共用的问题,以 96 CDI 为例,每一列的 i5 标签都是相同的,每一行的 i7 标签也都是相同的。当标签发生跳跃,形成新 i5 与 i7 组合时,这一组合产生的错误数据将无法被剔除。除此以外,组合型的样本标签需要将不同的 i5 与 i7 标签组合使用,一旦发生标签污染,就会引入假阳性。序列特异双端标签(Unique Dual Index–UDI)不存在标签共用问题,一对样本标签同时两两跳跃到另一段 DNA 片段上的概率几乎为零,因而可以更好的解决标签跳跃问题。


384 对序列唯一双端标签(384 UDI)满足高通量测序混样需求,同时有效应对标签错配,预混好的 384 UDI,方便操作,避免手动混合潜在的交叉污染风险 双端分子标签(Dual MBC)校正 PCR 与测序过程的假阳性,包括 PCR 早期引入的假阳性 优化的接头连接体系,大幅提高转化效率,生成高复杂度的文库,从微量的 FFEP 和液态活检样本中获得尽可能全面的信息 样本片段化的兼容性,兼容机械打断与酶切打断(后者在低频变异应用中表现更为出色) 样本的兼容性,FFPE、低质量的 FFPE, ctDNA 等样本均可采用同一操作流程 灵活的工作流程,既可以在一天内完整杂交、捕获,又可以过夜杂交 灵活的包装,包括自带磁珠的包装,方便订购 另外,安捷伦 SureSelect 酶切片段化试剂盒具有很好的 Tris/EDTA 浓度兼容性,基本无需稀释,即可对不同样本采用同一酶切程序


 长按识别二维码, 关注安捷伦视界
长按识别二维码, 关注安捷伦视界04-20 环亚生物
InnoQuant激光共聚焦全景玻片扫描在非小细胞肺癌肿瘤微环境研究中的应用04-20 环亚生物
拍出最真实最清晰的图片—InnoQuant助力生物组织荧光切片扫描和分析04-20 环亚生物
全景激光共聚焦扫描技术在生物学研究领域应用 ——荧光定量切片&芯片扫描学术讲座圆满落幕04-20 环亚生物
GLMY创想仪器丨河南铸锻年会展示优质光谱分析技术04-20
GLMY创想仪器丨亮相江苏东海高纯石英材料产业大会04-18
瑜伽裤有“毒”?Lululemon被曝添加“永久性化学品”:PFAS危害到底有多大?04-17
全自动DNA脉冲场电泳回收仪用于5' 转录组测序及CUT&RUN《Nature》-蚂蚁嗅觉基因选择性表达机制新发现04-17 环亚生物
超长测序文库新思路:Blue Pippin全自动大片段DNA脉冲场电泳回收仪用于Ultra Long 文库构建04-17 环亚生物
文献速递|Blue Pippin全自动DNA脉冲场电泳回收仪用于单分子蛋白组学识别技术开发04-17 APGBio
Pippin 系列全自动DNA片段回收仪用于GUIDE-seq2测序——《Nature》 CRISPR–Cas9 PAM变体研究04-17
展会预告 | 安东帕邀您共赴 Chinaplas 202604-17
免费试用70天!安东帕傅里叶变换红外光谱仪助力药企生产04-17 Anton Paar China
精酿人必看 | 如何让啤酒新鲜度“破局”?三大福利,抓紧来领!04-17 Anton Paar China
邀请函 | 密度黏度联合用户培训会@上海04-17 安东帕中国
展会邀请,欢迎理论指导04-17 上海棱光技术
GLMY创想仪器丨于杭州聚焦再生生物油脂高质量检测04-17
脂质研究必看:2400+产品目录,四大难题一网打尽04-16
第35届化学年会圆满落幕,Sigma-Aldrich®赋能化学,共启化学探索之旅04-16
与时俱进,与药典接轨:一文看懂离子对试剂怎么选04-16 Merck