从“已知修饰统计”到“未知修饰发现”
2026-03-25 15:39:18, GPT5.4 上海易算生物科技有限公司

北航刘超课题组近期发表了最新算法文献OpenSpec,致力于解决高卷积nDIA Astral数据的未知修饰发现能力。今天我们结合PTM发现这个难题的背景知识一起为大家进行一下解读。
要点速览 • PTM 研究的关键难点,不是“能不能测到常见修饰”,而是“能不能系统发现参数表之外的意外修饰”。 • Astral-DIA 带来更高速度和更深覆盖,但也让混叠谱图的去卷积成为开放修饰寻找的前提。 • OpenSpec 的核心逻辑是:先重构 pseudo-MS/MS,再做 DDA 开放搜索,最后回到 DIA-NN 混合谱库二轮搜索。 • 对真实项目来说,这种路线同时服务于生物学发现、实验质控、流程优化和队列级稳定定量。 |
导语
在很多蛋白质组学项目里,真正被稳定报告出来的,往往只是蛋白世界里“最容易看见”的那一层:未修饰肽段,以及少数早已写进搜索参数的常见修饰。可现实中的蛋白质并不是静态分子。它们在翻译之后会发生磷酸化、乙酰化、泛素化、糖基化、氧化、脱酰胺、焦谷氨酸化、二硫键形成等多种变化,样本处理过程中还可能引入烷基化副产物、氧化伪影、半胱氨酸相关副反应等质量偏移。换句话说,真正决定功能状态的,常常不是“这个蛋白在不在”,而是“它以什么修饰状态存在”。[1][2]
这也是为什么翻译后修饰(PTM)检测一直是质谱蛋白质组学中最有技术含量、也最有临床与转化价值的方向之一。修饰改变的不只是分子量,还会改变蛋白活性、亚细胞定位、互作网络、降解速率以及疾病相关通路状态。对于生物标志物发现、药物机制研究、工艺质控以及临床队列回溯分析来说,谁能更完整地看到修饰,谁就更接近真正的生物学解释。[1][11]
问题在于,“看到修饰” 和 “系统发现意外修饰” 是两件难度完全不同的事。前者依赖已知假设;后者要求算法面对更大的搜索空间、更复杂的谱图重叠以及更严格的错误控制。尤其是在 Orbitrap Astral 这样的新一代高通量平台上,DIA 数据拥有前所未有的速度、灵敏度和覆盖度,但也把“多肽共碎裂、谱图混叠、非预设修饰难发现”的老问题推到了更前台。[1][4][9]
近期,刘超课题组、杨静华课题组携手易算生物合作发表的 OpenSpec,正是针对这个痛点给出的一套新答案:先把 Astral-DIA 中复杂的混叠碎片信息重构为更接近单肽的 pseudo-MS/MS,再借助成熟的 DDA 开放式搜索能力发现意外修饰,最后回到 DIA混合谱库二轮搜索中获得更稳健的鉴定与定量结果。 这篇文章之所以值得关注,不仅因为它“多找到了几个修饰”,更因为它把“发现未知质量偏移”与“回到可定量的 DIA 工作流”真正打通了。[1]

图1 限制式搜索 vs 开放式搜索
一、为什么翻译后修饰是蛋白质组学真正的“高价值区”?
如果把基因组看作“蓝图”,蛋白质组更像“施工后的真实建筑”;而翻译后修饰,决定的是建筑是否通电、是否上锁、是否与别的系统联动。蛋白是否被磷酸化,决定某条信号通路是否被激活;赖氨酸是否乙酰化,可能影响染色质状态与转录调控;半胱氨酸是否形成二硫键、是否被进一步氧化,则常常牵连到氧化还原稳态、样本处理质量甚至蛋白构象稳定性。[1][11]
更重要的是,很多真正有解释力的现象,往往并不体现在蛋白总量上,而体现在proteoform / peptidoform 层面。也就是说,同一个蛋白可能总量不变,但它在不同处理组、不同疾病状态、不同细胞亚群中,表现为不同的修饰组合和不同的肽段状态。这也是为什么近年来从单细胞蛋白质组、单细胞组蛋白修饰到血浆糖蛋白组,越来越多工作把焦点从“多少蛋白”转向“多少功能状态”。2025 年的 sc-hPTM 工作就表明,单细胞层面的组蛋白修饰分析已经可以读出 67 种 peptidoform,并区分药物处理后的细胞异质性,这说明 PTM 研究正在从 bulk 样本快速向更精细的分辨率推进。[11]
当然,必须强调一个经常被忽略的事实:开放式修饰寻找发现的并不全是“真正的生物学 PTM”。 它也会发现样本处理造成的化学修饰、氧化伪影、脱酰胺、半胱氨酸副反应以及仪器/流程相关的偏移。看似“杂”,其实非常有价值。因为这既是生物学发现的入口,也是实验质控和流程优化的窗口。OpenSpec 在 IAA 与非 IAA 处理的对照中对半胱氨酸相关修饰的系统比较,恰恰说明了这一点:意外修饰发现不仅服务于生物学,也服务于方法学。[1]
二、质谱检测 PTM 的基础知识:为什么“知道有修饰”容易,“完整发现修饰”很难?
1)PTM 在质谱里本质上表现为什么?
对底物肽段来说,修饰最直接的后果是带来质量变化(mass shift),同时还可能改变液相保留时间、离子化效率和碎裂行为。因此,质谱识别 PTM 通常依赖四层信息同时成立:
第一层是 前体离子质量。修饰后的肽段相对未修饰肽段会出现特定 Δmass。
第二层是 MS/MS 碎片离子匹配。只有当 b/y 离子或其他诊断离子支持某个序列与某种修饰时,鉴定才更可信。
第三层是 修饰位点定位。同样的总质量偏移,可能落在不同氨基酸上;位点不清,生物学解释就会打折。
第四层是 定量与重现性。对科研和转化项目而言,发现只是第一步,后续必须能在多样本、重复间稳定量化。[1][2][3]
2)标准 bottom-up 流程怎么做?
在常规 bottom-up 蛋白质组学中,样本先裂解、变性、还原、烷基化,再经胰蛋白酶等消化成肽段,经液相分离后进入质谱。MS1 看到前体离子,MS2 负责碎裂与序列解析。对于 PTM 检测,研究者通常会在数据库搜索中设置固定修饰和可变修饰,例如半胱氨酸 Carbamidomethyl、甲硫氨酸 Oxidation、蛋白 N 端 Acetylation 等。[1][3]
这种策略对已知且高频的修饰非常有效,但也有天然上限:如果研究者事先不知道样本里有什么修饰,就不知道应该把哪些质量偏移写进参数;而一旦写得太多,搜索空间又会急剧膨胀,导致计算成本升高,罕见修饰的检测灵敏度反而下降。Unimod 数据库记录的修饰类型远不止常规软件界面推荐的几种,单靠“经验填参数”无法覆盖真实样本中的全部变化。[1]
3)DDA 为什么更适合传统开放搜索,DIA 为什么更难?
开放式搜索(Open Search)的基础思想并不新。以 MSFragger 为代表的定位感知开放搜索方法,已经证明可以在宽质量偏移空间里更高灵敏度地发现修饰肽段,并为修饰位点定位提供支持。[2] 但这套逻辑最初主要建立在 DDA 数据上:DDA 的单个 MS/MS 光谱通常来自较窄隔离窗,往往只包含一个主要肽段的碎片,谱图更“干净”。
而 DIA 不同。它会在更宽或连续设计的隔离窗中同时碎裂多个共洗脱前体,因此单张 MS/MS 里往往混有多个肽段的碎片峰。DIA 的优点是覆盖更全、重现性更强、缺失值更少;难点是谱图卷积更严重。也就是说,DIA 的原始数据本身更适合大规模定量,却天然不适合“拿来就做开放修饰搜索”。Spectronaut20引入的Probing search很好地解决了常见(40种左右)修饰的快速寻找,但对此之外的修饰就无能为力了。这正是过去几年相关算法反复攻克的问题。[1][3][5][6]
三、为什么 Astral-DIA 把“意外修饰寻找”推成了一个新问题?
Orbitrap Astral 的出现,改变的不只是扫描速度,而是整个数据分析问题的边界。2025 年针对 Astral 的多项研究显示,这一平台凭借高速度、高灵敏度与较高分辨率,使窄窗 DIA(nDIA)成为现实。nGlycoDIA 的工作明确写到,Astral 可提供 >200 Hz 的 MS/MS 扫描速度,并已被用于 40 分钟内完成血浆 N-糖蛋白组深度分析,累计鉴定超过 3000 个 unique glycoPSM、覆盖 181 个糖蛋白。[9] 在极微量蛋白质组场景中,Bubis 等人的研究则显示,Astral 在单细胞量级样本上量化到的蛋白数超过 Exploris 480 的 2 倍,且低丰度蛋白的定量 CV 更低。[4]
这些进步给 PTM 研究带来两个相反的结果。
一方面,更快、更深、更稳 的 DIA 数据让修饰发现拥有了更好的原始信号基础;
另一方面,数据量和复杂度同时上升,意味着如果算法还停留在“只认几种默认修饰”的老范式,大量高价值信息依然会被丢在数据里。
这也是近一年相关方法集体演化的原因。diaTracer 为 diaPASEF 数据做三维峰追踪,生成 precursor-resolved 的 pseudo-MS/MS,本质上是在为直接谱图解析“清场”;CHIMERYS 则把 DDA、DIA、PRM 都看成可被统一去卷积的谱图问题,通过保留时间、碎片强度预测和正则化线性回归来解释混叠碎片;DIA-BERT 利用大规模预训练 Transformer 直接增强 DIA 鉴定与定量;Carafe 则让谱库模型直接从 DIA 数据学习,而不是完全依赖 DDA 训练得到的先验。[5][6][7][8]
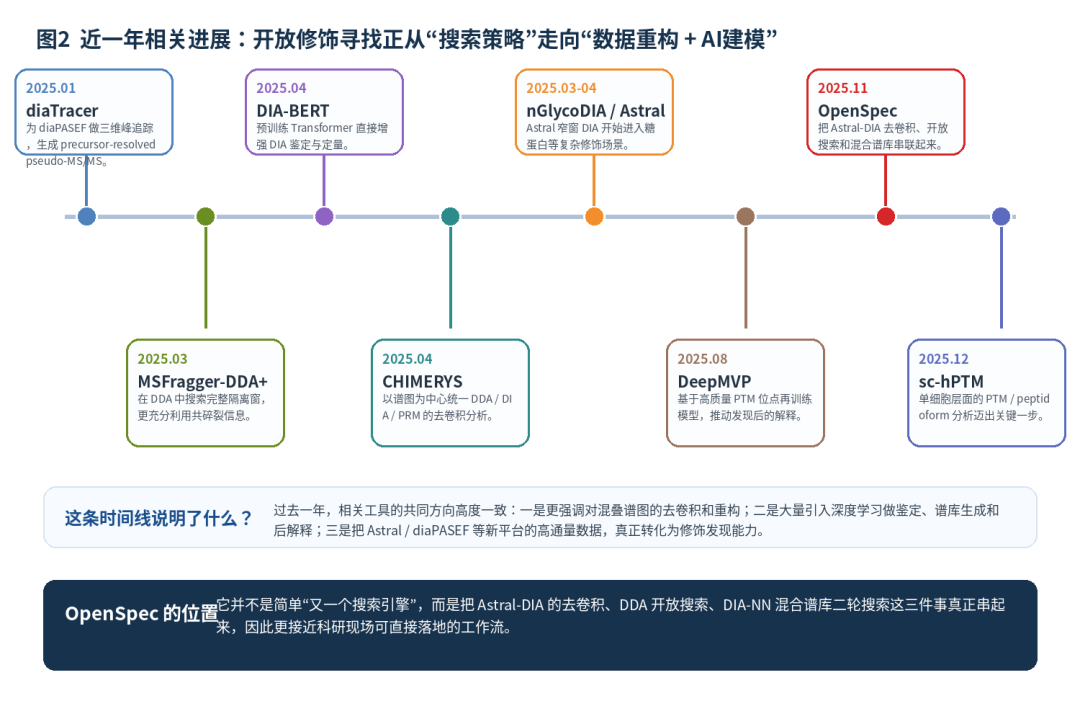
图2 近一年相关进展
近一年相关方法对比(面向开放修饰寻找的启发)
工作 | 数据/平台 | 核心思想 | 对开放修饰寻找的启发 | 年份 |
diaTracer | diaPASEF | 三维峰追踪,生成 precursor-resolved pseudo-MS/MS | 说明“先重构谱图,再谈深度发现”已经成为主线。 | 2025 |
CHIMERYS | DDA / DIA / PRM | 以谱图为中心统一去卷积,允许一张谱图解释多个肽段 | 证明混叠碎片不是噪音,而是可被建模利用的信息。 | 2025 |
DIA-BERT | DIA | 预训练 Transformer 增强鉴定与定量 | AI 开始直接进入 DIA 鉴定主流程。 | 2025 |
Carafe | DIA | 让谱库预测直接从 DIA 数据学习 | 为修饰肽后续进入谱库与稳定追踪提供思路。 | 2025 |
OpenSpec | Astral-DIA | 去卷积 + DDA 开放搜索 + 混合谱库二轮搜索 | 把“发现未知修饰”和“回到可定量 DIA”真正打通。 | 2025 |
如果把这些工作放在一起看,会发现一个很清晰的技术趋势:未来的 PTM 与开放修饰发现,不再只是“搜索引擎更快一点”,而是“先把复杂数据重构清楚,再让 AI、开放搜索和谱库定量形成闭环”。[1][5][6][7][8]
四、Open modification search 到底进化到哪一步了?
开放式修饰寻找过去常被理解为“给 precursor tolerance 放大一点”。今天来看,这个理解已经不够了。真正成熟的开放修饰发现,至少包含四个层级:
第一层:能发现质量偏移。
这是 open search 的入口,解决“参数表里没写的修饰怎么进来”。
第二层:能解释质量偏移。
也就是把一个 Δmass 与可能的修饰类型、位点和碎裂证据联系起来,而不是只给出一个模糊数值。
第三层:能回到稳定定量。
如果发现出的修饰无法在后续 DIA / library search 框架中被稳健追踪,那么科研价值会大打折扣。
第四层:能与实验流程和质控联动。
尤其对半胱氨酸、氧化还原相关修饰、糖基化以及样本处理敏感的项目,算法输出必须能反馈给样本制备策略。
OpenSpec 的亮点在于,它不是只做到第一层,而是把第一层到第三层串了起来,并在第四层上给出了实际示范。[1]
五、OpenSpec 做了什么?
一:先把 Astral-DIA 去卷积成 DDA-like 的 pseudo-MS/MS
作者首先不是直接拿 DIA 原始谱图做开放搜索,而是先做特征级别重构。具体来说,先提取 candidate precursor feature 与 candidate fragment feature;再用一个 Transformer-based precursor-fragment grouping model 评估它们是否来自同一条肽段,输出 0 到 1 的匹配分数;最后把高置信匹配的前体与碎片合并成 DDA-like 的 pseudo-MS/MS。[1]
这一步是整篇工作的地基。因为只要谱图还处于“多个共洗脱肽段缠在一起”的状态,开放搜索再强也很难真正发挥。OpenSpec 的策略与 diaTracer、CHIMERYS 的共同点,都是先承认 DIA 光谱天然混叠,再通过建模把它“拆开”;不同点在于,OpenSpec 的整个后续设计是为 Astral-DIA 上的意外修饰发现 服务的。[1][5][6]
二:用成熟的 DDA 开放搜索引擎先把“意外修饰名单”找出来
生成 pseudo-MS/MS 之后,OpenSpec 采用 pFind3 做开放搜索,利用 Unimod 修饰空间去发现数据里高丰度的 unexpected modifications。文中随后把最丰的 6 类修饰定义为后续分析的高丰度修饰候选,用于第一轮 DIA-NN 搜索和后续混合谱库构建。[1]
这一步非常聪明。因为它没有试图“重新发明一个包含所有功能的新搜索引擎”,而是把 DDA 生态里已经很成熟的开放搜索能力借过来。换句话说,OpenSpec 的创新不是替代现有工具,而是重新安排工具之间的顺序:先去卷积、再开放搜索、再回到 DIA 搜索框架里量化。
三:把发现结果回灌到 DIA软件的混合谱库二轮搜索
这一步是 OpenSpec 真正区别于“只做 open search 然后停下”的地方。作者先用第一轮 DIA-NN 在设定高丰度修饰的条件下得到 library-free 结果,再把这些结果与开放搜索得到的修饰肽段合并,构建一个包含 unexpected modifications 的 hybrid spectral library,最后再进行第二轮 DIA-NN 搜索。[1]
为什么这一招重要?因为开放搜索只解决了“发现”,但真实项目还需要“稳定量化”。如果修饰肽段没有进入混合谱库,它们在保留时间、碎片强度和竞争关系上的建模就不够完整,后续搜索很容易漏掉。OpenSpec 通过二轮搜索,把“发现修饰”变成“可被 DIA 工作流持续追踪的修饰”。这是它最有应用价值的设计之一。[1]
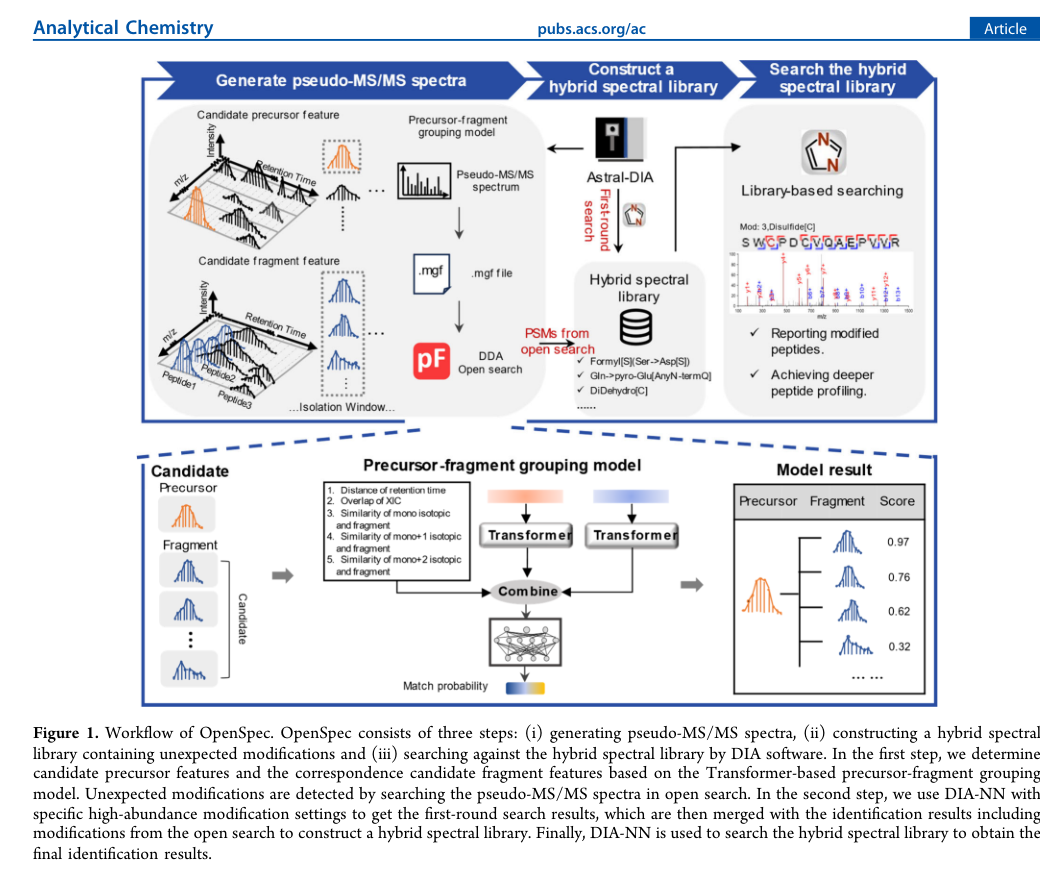
OpenSpec 原文工作流图

图3 OpenSpec 关键结果
六、这篇论文的核心结果,应该怎么理解?
1)合成肽验证:证明 pseudo-MS/MS 不是“看起来像”,而是真能保留修饰信息
作者设计了 20 条合成肽,其中 10 条带一个修饰,10 条带两个修饰,用来模拟不同 unexpected modifications。在不预设所有修饰的情况下,单轮 DIA-NN 即便设置多组参数,也无法完整报告全部 20 条目标肽;在 5% peptidoform FDR 下只能报告 7 条,而且没有一条是双修饰肽。相反,OpenSpec 生成的 pseudo-MS/MS 经 pFind3 开放搜索后,可以成功找回这些修饰肽;再把它们写入混合谱库并做第二轮 DIA-NN 搜索后,20 条合成修饰肽都能在 5% peptidoform FDR 下被报告。[1]
这说明两个问题:第一,OpenSpec 的去卷积足够保留修饰信息;第二,单轮搜索不够,发现结果必须回到混合谱库里完成“再搜索”。
2)与 DIA-Umpire 对比:说明不是所有 pseudo-MS/MS 都一样好
论文还把 OpenSpec 的 pseudo-MS/MS 生成模块与经典 DIA-Umpire 导出流程做了对比。在相同后续搜索框架下,作者报告 OpenSpec 生成的 pseudo-MS/MS 经 pFind3 搜索后,识别到的肽段数量约比 DIA-Umpire 导出结果高 50%,且在修饰肽找回上更完整。[1]
这意味着,对于 Astral-DIA 这样的高速度、高复杂度数据,并不是把旧有去卷积工具原样搬过来就够了。新平台需要新一代的特征配对和谱图重构策略。 这恰恰解释了为什么 OpenSpec 选择用 Transformer 做 precursor-fragment grouping,而不是只依赖传统的相关系数与人工阈值。[1]
3)DIA-DDA 配对验证:证明 OpenSpec 找到的不是“幻觉修饰”
在同一样本的 DIA / DDA 配对数据中,OpenSpec 在 DIA 数据里找出的高丰度修饰,与 DDA 的开放搜索结果在修饰类型和排序上高度一致。文中列出的 top 6 修饰包括:Carbamidomethyl[C]、Formyl[S](Ser -> Asp[S])、Deamidated[N]、Gln -> pyro-Glu[AnyN-termQ]、Acetyl[ProteinN-term] 和 Oxidation[M]。作者还比较了同一修饰肽在 DIA 和 DDA 中的保留时间差,大多数都在 5 秒以内。[1]
这组结果很重要,因为它告诉我们:OpenSpec 并不是在 DIA 中“想象出”一些质量偏移,而是能在与 DDA 相互印证的前提下,稳定发现高丰度意外修饰。
4)深度提升:发现修饰之后,整体前体鉴定也更深
在 DIA-DDA 配对数据上,作者报告:
- 只使用常规变量修饰设置时,三次重复的平均前体数为 178,123;
- 根据 OpenSpec 找到的 top 6 高丰度修饰来优化 DIA-NN 参数后,平均前体数升到 179,479;
- 再导入混合谱库进行第二轮搜索后,平均前体数达到 188,235。[1]
按文中数值计算,混合谱库二轮搜索相对于常规设置大约提升 5.7%。对已经非常深的 Astral-DIA 数据来说,这不是一个小幅度。更关键的是,提升并非来自“放松阈值”,而是来自更准确地把修饰肽带回了可竞争的搜索框架。
5)IAA / 非 IAA 对照:把“意外修饰发现”变成样本制备优化工具
OpenSpec 还有一个很实用的展示:作者对同一 293T 样本分别做了加 IAA 与不加 IAA 的处理,用来观察半胱氨酸相关的 unexpected modifications。结果显示,在不加 IAA 的样本里,Disulfide[C] 是最丰富的半胱氨酸相关修饰;而在 IAA 处理 样本中,Carbamidomethyl[C] 占绝对优势,其他修饰显著下降。[1]
更值得注意的是定量结果:IAA 处理组平均鉴定到 154,993 个前体,其中含半胱氨酸的前体为 23,238;不加 IAA 组平均仅 127,506 个前体,含半胱氨酸前体只有 1,991 个。也就是说,IAA 处理不仅提高了整体可识别深度,还使含半胱氨酸前体的可识别数量达到未处理组的 约 11.7 倍。[1]
这段结果非常适合公众号传播,因为它直接回答了很多实验室日常会遇到的一个问题:“烷基化到底有没有必要?” OpenSpec 给出的答案不是经验判断,而是数据驱动的修饰景观比较。
七、OpenSpec 的真正优点,不只是“能开放搜索”
把整篇论文浓缩成一句话,OpenSpec 的真正优势有六点。
第一,它抓住了 Astral-DIA 的核心矛盾。
Astral 带来了更快、更深、更稳的 DIA,但也让非预设修饰的发现变得更迫切。OpenSpec 没有回避 DIA 的混叠问题,而是正面解决它。[1][4][9]
第二,它把 AI 用在了最该用的地方。
不是为了“做一个更炫的黑盒”,而是用 Transformer 去判断 precursor 和 fragment 是否应归属于同一肽段,这恰好是谱图去卷积最困难、最值得建模的环节。[1]
第三,它兼容现有主流生态。
OpenSpec 并未替代 DIA-NN 或 pFind3,而是把两者组合成一个更强的流程。这意味着实验室不需要推翻既有分析习惯,就能把 unexpected modifications 纳入更成熟的 DIA 分析框架。[1]
第四,它把“发现”与“定量”闭环了。
很多开放搜索工作停留在“发现一堆质量偏移”;OpenSpec 则继续往前走,用混合谱库把这些修饰带回稳定定量体系。对真实项目而言,这一步比“发现更多修饰”本身更重要。[1]
第五,它兼顾了生物学发现与方法学质控。
IAA 对照实验说明,unexpected modifications 的价值并不只在新生物学,也在实验优化、流程排错、样本质量评估。[1]
第六,它没有额外消耗临床样本。
作者在结论里明确指出,OpenSpec 可以与现有主流搜索引擎结合,在不额外消耗临床样本的情况下报告意外修饰,这对珍贵队列和回顾性数据再分析尤其有吸引力。[1]
当然,OpenSpec 也有边界。就目前论文而言,它主要聚焦于 Orbitrap Astral 产生的 DIA 数据;对其他平台和更多修饰类型的泛化,还需要后续扩展。另一方面,开放修饰发现永远需要谨慎解释:一个质量偏移被发现,并不自动等于该修饰具有明确生物学功能,仍然需要结合位点定位、重复一致性、样本处理背景和独立验证来判断。[1][10]
八、OpenSpec 的价值
OpenSpec的编写目的放在近一年方法学演化的语境里,它其实对应了三个行业级趋势。
趋势一:质谱平台升级后,数据分析成为新的主战场
Astral、diaPASEF 等平台正在把原始数据质量推向新高度,但真正能否转化为修饰发现能力,取决于后端算法。diaTracer、CHIMERYS、DIA-BERT、Carafe、OpenSpec 这些工作,都是在回答同一个问题:高速高覆盖数据,如何不被传统分析范式“浪费掉”?[5][6][7][8][9]
趋势二:AI 不再是外围插件,而是进入谱图重构、鉴定和谱库学习核心环节
DIA-BERT 用预训练 Transformer 直接增强 DIA 鉴定;Carafe 让谱库预测直接从 DIA 数据学习;OpenSpec 则把 Transformer 放在 precursor-fragment 分组这个关键问题上。可以预见,接下来 PTM 发现的竞争,不只是“谁的数据库更大”,而是“谁能更准确地在复杂数据中重建真正的肽段证据”。[6][7][8]
趋势三:错误控制将变得越来越重要
2025 年针对 entrapment 的 FDR 评估研究提醒大家,DIA 分析中的错误控制仍然是一个需要认真面对的问题,尤其在复杂或超低输入数据上,没有哪种 DIA 搜索工具能在所有场景下都稳定控制 FDR。[10] 这也反过来解释了为什么 OpenSpec 采用“去卷积 + 开放搜索 + 混合谱库二轮搜索”的组合路线更有现实意义:它没有试图用单一环节解决所有问题,而是尽量把发现、验证和定量拆开做对。
九、从论文到方法学落地
易算观察|把方法学真正落地,比“会做 open search”更重要 • 易算生物当前覆盖 Astral / 4D tims DIA 质谱技术服务、低丰度蛋白富集、各类翻译后修饰富集、多组学定制实验及分析,并布局 OmicsCloud、悟空云、EasyDIA、GlycAP 等数据分析平台。 • 这意味着对需要从样本制备、实验执行到算法与结果解释一体化推进的项目,易算是“方法 + 服务 + 平台”的组合伙伴,而不是单纯的检测供应商。 |
这对科研工作者意味着什么?很简单:
当行业开始进入“新平台 + 新算法 + 新修饰发现”的阶段,真正稀缺的不再只是仪器 access,而是从样本策略、实验执行、算法选择到结果解释的一体化能力。对希望做深度发现蛋白质组、修饰组、低丰度蛋白挖掘或临床队列回溯分析的团队来说,谁能把这几段链条接起来,谁就更容易把数据转化成可发表、可复现、可解释的结果。[12]
从这个意义上说,OpenSpec 不只是一次论文合作,更像是一个信号:下一阶段的质谱服务竞争,会越来越从“做实验”升级到“做方法 + 做算法 + 做平台”。 这也正是易算这类兼具实验与计算能力的公司值得被更多用户关注的原因。
结语:PTM 研究正在进入“先发现、再解释、再定量”的新阶段
过去很多 PTM 项目,核心难点是“测不出来”;今天,随着 Astral、窄窗 DIA、AI 去卷积与开放搜索的发展,难点正在变成“怎样把未知修饰从复杂数据里可信地找出来,并且让它进入稳定定量框架”。OpenSpec 的价值,就在于它给出了一个非常完整的范式答案:先重构,再发现;先开放,再回灌;先把 unexpected modifications 看见,再把它们变成可被追踪的 peptidoform。[1]
对于科研端,这意味着更多机制线索、更少参数盲区;
对于方法学端,这意味着更好的样本制备反馈和流程优化;
对于产业端,这意味着新平台红利终于有机会真正转化为服务能力和数据价值。
如果说传统蛋白质组学解决的是“我们测到了什么蛋白”,那么接下来真正决定竞争力的问题会是:我们有没有能力从同一份数据里,发现那些过去根本没有被写进参数表的修饰状态? OpenSpec 给出的回答,值得这个领域认真对待。
参考文献
[1] Tang M, Sun Z, Shen C, et al. OpenSpec Enables Detecting Unexpected Modifications from Proteomics Data Generated by Orbitrap Astral Mass Spectrometer. *Analytical Chemistry*. 2025.
[2] Yu F, Teo GC, Kong AT, et al. Identification of modified peptides using localization-aware open search. *Nature Communications*. 2020;11:4065.
[3] Lou R, Shui W. Acquisition and Analysis of DIA-Based Proteomic Data. *Molecular & Cellular Proteomics*. 2024;23(2):100712.
[4] Bubis JA, Arrey TN, Damoc E, et al. Challenging the Astral mass analyzer to quantify up to 5,300 proteins per single cell at unseen accuracy to uncover cellular heterogeneity. *Nature Methods*. 2025;22:510-519.
[5] Li K, Teo GC, Yang KL, et al. diaTracer enables spectrum-centric analysis of diaPASEF proteomics data. *Nature Communications*. 2025;16:95.
[6] Frejno M, Berger MT, Bronshtein I, et al. Unifying the analysis of bottom-up proteomics data with CHIMERYS. *Nature Methods*. 2025;22:1017-1027.
[7] Liu Z, Liu P, Sun Y, et al. DIA-BERT: pre-trained end-to-end transformer models for enhanced DIA proteomics data analysis. *Nature Communications*. 2025;16:3530.
[8] Wen B, MacLean BX, Berg MD, et al. Carafe enables high quality in silico spectral library generation for data-independent acquisition proteomics. *Nature Communications*. 2025;16:9815.
[9] Jager S, Zeller M, Pashkova A, et al. In-depth plasma N-glycoproteome profiling using narrow-window data-independent acquisition on the Orbitrap Astral mass spectrometer. *Nature Communications*. 2025;16:2497.
[10] Wen B, Freestone J, Riffle M, et al. Assessment of false discovery rate control in tandem mass spectrometry analysis using entrapment. *Nature Methods*. 2025;22:1454-1463.
[11] Cutler R, Corveleyn L, Ctortecka C, et al. Mass spectrometry-based profiling of single-cell histone post-translational modifications to dissect chromatin heterogeneity. *Nature Communications*. 2025;16:11100.
[12] Omicsolution 易算生物官网
07-23
化学实验基石:如何制作一条“靠谱”的标准曲线?(下)07-23
连华故事·第5期 | 信得过,所以一直用07-23
活动过半提醒 |618+818双擎守护 购买产品即送豪礼07-23
国资委46号令施行:科学仪器自主创新,更要经得起验证07-22
标称65%的探测效率,实测却只剩10%多:SPAD单光子探测效率,差距到底藏在哪?07-21 光电传感器量测
NK细胞扩增"利器"—— GMP级基因改造K562滋养层细胞07-21
CGT产品RCV风险怎么控?CDE最新征求意见稿深度解读07-21
广拓新境,再攀“高度”|Altura 生物分析版图再扩容07-21 安捷伦科技
PFAS 检测再升级:安捷伦创新方案破解生物固体样本分析难题07-21 安捷伦科技
ACT-UR 支持项目登上《Nature》——安捷伦先进分析技术助力揭示蜂王发育的“建筑密码”07-21 安捷伦科技
【专题会议】聚焦 Vaya 手持拉曼,安捷伦助力原辅料快速、合规、精准入场质检07-21 安捷伦科技
科学搬家 vs 普通搬家:深度拆解实验室搬迁服务的分水岭07-21 安捷伦科技
半导体超痕量分析新选择 | 安捷伦 PFA‑ST MicroFlow 微流量雾化器07-21 安捷伦科技
制备色谱放大务必关注背压 —— 填料粒径与流动相组成对背压的影响07-21 绿绵科技
邀请函 | 2026(第十二届)全国香料香精技术交流年会07-20 大昌华嘉
网络研讨会 | 基于EOF和AOF分析的水基质中PFAS筛查07-20 大昌华嘉
邀请函 | 第十届细胞外囊泡合规与临床应用大会07-20 大昌华嘉
网络研讨会 | 新一代NTA技术助力细胞外囊泡质量评估与工艺开发07-20 大昌华嘉
网络讲堂 | 7月24日 核磁共振分析仪在食品科学中的应用,诚邀莅临!07-20 哈希公司