利用人工智能突破一维核磁共振光谱的局限,实现自动化分子结构解析
2026-02-04 15:52:49, ACDLabs 朱秦 Advanced Chemistry Development, Inc. (ACD/Labs)



核心突破


AI攻克化学空间组合爆炸难题
传统一维NMR结构解析面临两大瓶颈:一是化学空间的爆炸式增长,分子原子数增加会导致可能结构数量呈指数级飙升;二是缺乏高效的自动化工具,需结合二维NMR、高分辨质谱等额外数据,依赖复杂的人工分析或计算机辅助结构解析 (CASE) 方法。
斯坦福大学团队的创新框架通过三大核心设计实现突破, 且在初代框架基础上完成关键升级:
1. Transforming架构+分子指纹预训练
奠定高准确率基础
研究团队借鉴自然语言处理的思路,设计了基于Transformer的深度学习架构。首先通过“子结构-结构”预训练任务优化模型——利用Morgan指纹 (一种能高效表征分子子结构的二进制向量) 作为输入,训练模型重构分子的SMILES字符串。

图2:子结构到结构任务的结果。(A) 错误预测的比例以及错误预测与目标分子的平均最大塔尼莫托相似度 (MTS) 随重原子数量变化的情况。虚线是每个量在不同大小范围内的简单移动平均值。(B) 与目标分子的MTS分布,按不同重原子数量范围进行分解。(C) 子结构到结构转换器正确预测的分子示例,所有分子都有40个重原子且元素组成各不相同。
图A中蓝色点代表预测错误率,橙色三角形代表平均最大塔尼莫托相似度 (MTS)。40个重原子分子的预训练模型准确率仍达88.8%,错误预测与正确结构的相似度超99.2%,展现出极强的泛化能力。
最新框架中,斯坦福大学团队将训练数据扩展至PubChem数据库的8800万个分子样本上,该预训练模型表现惊人:对于含40个重原子的分子,前15个预测结果中正确结构的准确率达97.8%。即便分子尺寸增大,模型准确率仅轻微下降,展现出对复杂化学空间的强大泛化能力。这得益于Morgan指纹的强大表征能力——半径为2、8192位的指纹能区分测试集中近150万个分子中的绝大多数,为模型提供了近乎唯一的序列表征。
2. 多任务框架直接“谱图-结构”转换
无需额外信息
将预训练好的Transformer模型集成到多任务框架中,实现两大核心功能:一是“谱图-结构”转换,直接从¹H和¹³C NMR光谱生成分子完整结构;二是“谱图-子结构”预测,同步识别分子中的关键子结构片段。
整个框架无需分子式、已知子结构等额外信息,仅需对光谱进行最小化预处理。在含40个重原子的分子测试集中,前15个预测结果的结构准确率达55.2%——要知道,该化学空间的分子数量已超过1030种,这一结果意味着AI成功突破了组合爆炸增长的限制。

图3:谱图到结构任务的结果。(A) 错误预测的比例以及错误预测与目标分子的平均最大塔尼莫托相似度(MTS)随重原子数量变化的情况。虚线是每个数量在不同大小范围内的简单移动平均值。(B) 在不同重原子数量范围内分解后,与目标分子的MTS分布。(C) 多任务模型正确预测的分子示例及其¹H NMR谱图。所有展示的体系重原子数量均在35-40之间。
3. 单光谱也能高效解析
适配实验场景需求
实际科研中,部分场景下获取¹³C NMR光谱可能受仪器限制。研究发现,该框架仅使用¹H NMR光谱时,结构解析准确率仍可达46.6%,子结构预测的F1分数达0.81,说明¹H NMR光谱中蕴含的连接性信息已足够支撑大部分结构解析需求,而¹³C NMR主要起到辅助验证作用。

更重要的是,该框架可通过少量实验数据微调适配实际场景:仅用50组实验NMR光谱进行微调后,对实验数据的结构解析准确率达20%,同时保持对模拟光谱的高准确率(54.6%vs微调前55.2%),解决了理论模型与实验数据脱节的关键问题。

图4:两个正确预测示例的模拟1H NMR光谱与实验1H NMR光谱对比,其中模拟光谱由ACD/NMR Predictors生成,实验光谱来自BMRB,并根据我们的工作流程进行了预处理。
性能表现
多维度验证可靠性
1、结构解析准确率领先,错误预测仍具参考价值
对于含20个重原子的分子,框架准确率高达70%;即便对于40个重原子的超大分子,准确率仍保持在10%——考虑到从20到40个重原子,化学空间规模从1015增长到 1036 (增加21个数量级),这样的准确率下降幅度已远低于预期。
值得注意的是,即便模型预测错误,其结果与正确结构的相似度也很高:所有错误预测的平均塔尼莫托相似度 (Tanimoto similarity) 达0.59,70.5%的错误预测相似度超过0.5,仅在个别键或原子上存在差异,仍能为实验人员提供重要参考。
2、子结构预测高置信度,辅助快速验证
框架的子结构预测功能表现同样出色:98.2%的子结构预测结果置信度高于0.9或低于0.1,其中预测不存在的子结构准确率达99.8%,预测存在的子结构准确率达96.5%。这种高置信度的子结构识别,可帮助研究人员快速验证结构解析结果,进一步加速研究进程。
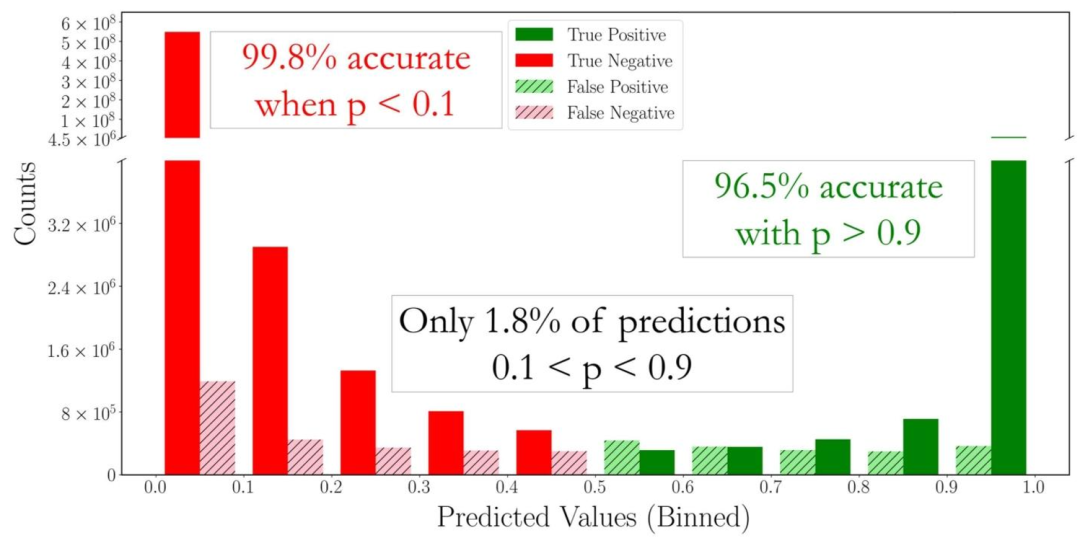
图10:根据预测概率以及真阳性、假阳性、真阴性、假阴性划分的多任务模型子结构预测性能。
模型预测结果集中在高置信区间(p>0.9或p<0.1),仅1.8%的预测处于模糊区间(0.1<p<0.9),体现了预测的可靠性。
3、全元素覆盖,适配复杂分子体系
该框架支持有机化学中常见的12种元素,包括传统方法难以处理的P、Si、B及卤素等。其中,含S、F、Cl等元素的分子解析准确率超过46%,即便对于训练集中占比不足2%的Si、B、P等元素,准确率也能维持在20%以上,展现出强大的元素泛化能力。
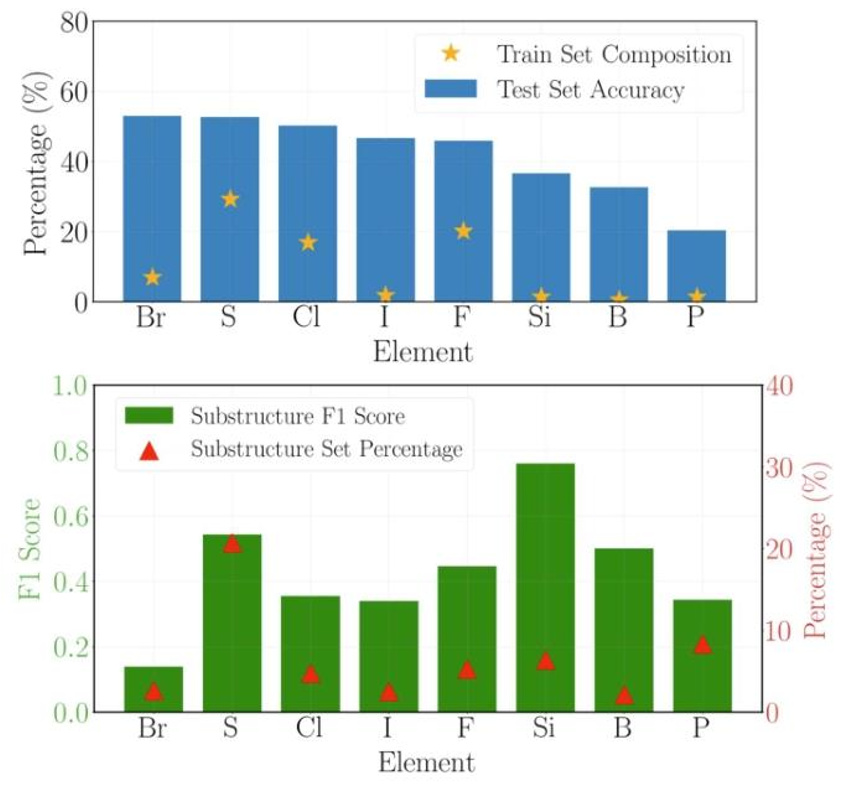
图4:(上)多任务模型测试集的结构预测准确率按元素划分,其中准确率是指包含该元素的体系中模型预测正确的百分比。(下)测试集的子结构预测准确率按元素划分,其中准确率以F1分数表示,子结构集占比是指整个子结构集中包含特定元素的子结构所占的比例。
为何P元素的预测难度更高?研究发现,63%的P原子为5价,37%为3价,成键方式的多样性导致其分子环境更复杂(含P子结构达237种),而Si和B分别固定为4价和3价,模型可通过数据集丰富的C、N 原子类比学习,泛化效果更优。
应用价值
加速化学发现全流程
该研究突破了一维NMR光谱的应用局限,为化学研究带来多重变革:
降低实验成本:无需依赖二维NMR、高分辨质谱等昂贵实验数据,仅用常规一维NMR就能完成结构解析,大幅降低实验门槛。
提升研究效率:模型生成15个预测结果仅需0.8秒(GPU)或2.8秒(CPU),能快速缩小化学搜索空间,将原本数天甚至数周的解析工作缩短至分钟级。
拓展应用场景:可广泛应用于有机合成、天然产物分离、药物研发等领域,尤其适合野外实验、小型实验室等缺乏高端仪器的场景。
互补现有工具:可与CASE等传统方法协同使用,形成更灵活高效的结构解析工具链,推动自动化化学研究的发展。
未来展望
从模拟到实验,从平面结构到立体化学
目前,该框架已在模拟数据和少量实验数据中充分验证了有效性。研究团队明确指出,尽管从一维核磁共振光谱实现完全从头结构解析仍面临两大核心挑战——一是立体化学测定(如区分对映异构体),二是弥合模拟数据与实验数据的差距,但这一基于Transformer架构的多任务框架,依旧为迈向全自动、大规模结构解析奠定了关键基础。
这一系列研究突破再次证明,AI正在重构化学研究的范式——从初代框架实现19个重原子分子的高效解析,到最新框架突破40个重原子的限制,深度学习与传统光谱技术的结合,正在让曾经看似“无解”的化学空间组合爆炸难题,逐渐找到高效破解之道。
点击文末
阅读原文
即可阅读原文章
往期推荐

基于CASE-DFT对浅黄枝衣中质子缺失型Chlorodepsidones的结构解析及Flavicansone的结构修订

06-03
【活动信息】马尔文帕纳科即将亮相第19届药机展PMEC202606-03 马尔文帕纳科
6月25日-成都-马尔文帕纳科多维材料表征技术研讨会06-03
【热点应用】当晶体学遇上无标记动力学:GCI技术如何助力片段药物发现的“去噪”与“加速”06-03 马尔文帕纳科
6月30日-海口-马尔文帕纳科多维材料表征技术研讨会06-03
科研利器 | Thorlabs保偏光纤跳线全家族:从高消光比到“加热对准”黑科技06-03 韵翔光电
别再只盯着色谱柱了!GC 分析问题可能藏在进样口密封件里06-03 安捷伦科技
从看不见的干扰到看得见的结果——Agilent J&W HP-5Q 色谱柱,让 GC/MS 分析更可靠06-03 安捷伦科技
618-柱压高怎么办?溶剂管理有妙招06-03 安捷伦科技
液相色谱柱堵塞频发?保护柱帮你守住分析生命线06-03 安捷伦科技
无人值守,高效快速|助力检测“泡药杨梅”中防腐剂与甜味剂06-03 安捷伦科技
打破极性歧视!PIDESI 双电离技术赋能原位质谱成像06-03 安捷伦科技
你的 GC,还在“慢跑”吗?06-03 安捷伦科技
化繁为简 智谱新篇 | 安捷伦新一代 ICP-MS/MS 与气相色谱系统正式登陆中国06-03 安捷伦科技
安全提效|精细化工反应风险评估解决方案 联购特惠来袭06-03 仰仪科技
水质农残管控|水中草甘膦残留检测测定方法06-03 科诺美Chromai
活动预告|科诺美邀您共聚天津大学第二十六届仪器设备展示会06-03 科诺美Chromai
聚力前行 悦享山海|纽迈舟山两日团建圆满收官!06-02 纽迈分析
不止均质精细!大批量杨梅抽检,靠什么稳住检测数据?06-02 一韦仪器
晶泰科技赋能希格生科再获IND:强活性 pan-TEAD抑制剂SIGX2649临床潜力凸显,AI+类器官模式持续验证06-02