法规解读 | 欧盟最新 《药品、活性物质、辅料和内包材灭菌指南》划重点
2023-02-15 14:18:55, 盛萍 Merck工艺解决方案

2019年3月8日,EMA发布了新的《药品、活性物质、辅料和内包材灭菌指导原则》 。该指南取代了原“灭菌方法决策树”文件(CPMP/QWP/054/98),为无菌产品选择合适的灭菌方法提供了依据和指导。
该文件将于2019年10月1日生效。

指南条文全英文?
当然也可以是简单易懂的,
为了增强
大家对《药品、活性物质、辅料和内包材灭菌指南》的理解,
今天小编给您划重点,
一起从无菌制药企业角度入手,
来解读欧盟这篇灭菌指南。
这个指南分为7个章节,前3个章节分别介绍了指南的背景、适用的范围以及法规依据。
第4个章节介绍了无菌制剂及无菌组分的生产要求和蒸汽灭菌、干热灭菌、电离辐射灭菌、气体灭菌、无菌过滤、无菌处理等不同灭菌方式的验证要求。
第5个章节介绍了溶液剂型产品,非溶液剂型、半固体或干粉产品和容器的灭菌方法选择决策树。
第6、7个章节分别是术语定义和参考文献。本文对该指南的重点章节解读如下:
通用要求
• 应在所有灭菌工艺前规定微生物负荷控制水平。高微生物负荷接受标准不应以灭菌工艺的能力或灭菌前任何微生物负荷减少步骤为依据。
• 为了确保制剂成品中的细菌内毒素水平可以接受,配方成分中的微生物污染应尽可能减少。如果相关的话,应提供配方组分和溶液中内毒素和微生物负荷的质量限度标准。
• 在生产最终产品过程中,所有与成品或与成品中的任何成分(活性物质或中间产品)接触的的过滤器应予以说明,并应在质量文档中提供表3中所述的信息。
• 如果使用二级包材(例如用于输液袋的二级袋或用于保持容器外部无菌的泡罩)对药品提供特定的保护,则应描述包装过程,包括风险评估,因为它可能影响成品的无菌性;例如,一级和二级容器之间的残留水分。
• 应提供有关何时执行包装步骤(灭菌之前或之后)以及采用的任何无菌技术的信息。应从微生物学的角度来建议合理的的工艺。如果二级包材的使用意味着对成品进行额外的灭菌,那么应说明关于无菌保证和对成品质量的任何潜在影响。
蒸汽灭菌
• 所有蒸汽灭菌工艺都要求F0 ≥ 8分钟且最低工艺温度≥110 °C。
• 如果使用欧洲药典5.1.1的参考条件(≥121°C,所有单位均为≥15分钟)进行灭菌,灭菌周期的验证数据无需在质量文档中提交。
• 如果使用用F0作为监测工艺杀灭力的额外控制, 则应说明 F0, 并由温度传感器测量的最低温度来确定F0。
• 在保温阶段,在成品温度低于115°C的情况下进行蒸汽灭菌是一种例外情况,应科学证明,并由表1中所述的额外数据来支持。如果在确定F0时包括低于110°C的温度(在加热和冷却过程中),则应说明这一点。
• 微生物负荷限制应符合任何灭菌前使微生物负荷减少的工艺能力(例如过滤)。对于水溶液,表1中规定的限值适用于原料药和制剂,无需进一步的说明。在规定的水平上控制微生物负荷的其他测试方法和限值应予以说明。
表1 蒸汽灭菌和无菌后热处理的程序以及质量文档中要求的相应数据

干热灭菌
• 使用欧洲药典5.1.1(至少160°C,至少2小时)的参考条件进行灭菌,不要求在质量文档中提交灭菌程序的验证数据。对于时间和/或温度低于药典中参考条件的灭菌循环,应提供灭菌程序的物理和生物验证,以证明SAL≤10^-6,如欧洲药典5.1.1所述。
• 对于注射用成品配方的最大生物负荷限制为100 CFU/100 g或100 CFU/100 ml是可以接受的,无需进一步证明。
• 对于不用于注射给药的活性物质和成品,最大总生物负荷限制为10 CFU/g或10 CFU/ml是可以接受的 ,无需进一步基于风险进行评估。在规定的水平上控制生物负荷的其他测试方法和限度应说明理由。对于空容器,还应确定合理的微生物负荷限度。
电离辐射灭菌
• 对于这种灭菌方法,参考吸收的剂量为≥25kgy。若经过说明和验证,其他剂量可可能用于达到SAL≤10−6。
气体灭菌
• 一般来说,只有在没有其他灭菌方法的情况下,气体灭菌才是可接受的。
• 它主要用于包装材料和设备的灭菌,因此只包含在包材的决策树中。
• 确保气体残留物或相关转化副产品的浓度低于使用成品时可能产生有毒影响的浓度。应证明清洗工艺的有效性。
• 应提供仪器说明、拟使用气体的定量数据、灭菌前的生物负荷、暴露于气体中的时间、灭菌周期每个步骤之前和期间的温度和湿度,以及(如适用)清除任何有毒气体残留物的条件。
• 应通过无菌测试定期检查每批工艺的有效性,确认工艺参数和生物指标均在其接受标准内。参数放行不适用于气体灭菌(根据欧洲药典第5.1.1章)。
• ETO是一种剧毒气体。只有在没有其他灭菌方法的情况下,ETO灭菌才是可接受的。
• 应按照ICH M7“药物中DNA反应性(致突变)杂质的评估和控制以降低潜在致癌风险”的要求进行评估,除非相关产品不在该指南的范围内。对于ICH M7范围以外的产品,对剧毒杂质,申请人应根据ICH M7或表2中规定的接受标准(以最合适的为准)规定限度。
表2 在ICH M7限度不适用的情况下,乙烯灭菌产生的有毒气体残留物阈值

除菌过滤
• 已灭菌过滤器的完整性应在使用前通过测试进行验证,除非另有说明和验证,并应在使用后立即通过在线测试进行确认。
• 对于常规商业生产,应在无菌过滤前立即对散装溶液进行生物负荷测试。
• 在大多数情况下,生物负荷测试可接受不超过10 CFU/100 ml(TAMC)的限制。
• 如果添加预滤器只是为了预防,而不是因为未过滤的中间溶液具有更高的生物负荷,则此限度也适用于预滤器之前,并且从GMP的角度强烈推荐。
• 预过滤前的生物负荷限值高于10 CFU/100 ml可能是可以接受的,如果这是由于已知的具有固有微生物污染的起始材料。在这种情况下,应证明在最后一次过滤之前,第一个过滤器能够达到10 CFU/100 ml的生物负荷。
• 开始中间溶液的制备和无菌过滤之间的最长时间应予以说明,应最小化,并由数据提供适当支持。过滤时间超过24小时应予以说明。
• 如果在24小时内未将无菌过滤后的溶液装入最终产品容器中,除非另有说明,应在灌装前立即重复无菌过滤。在保持时间之后的任何进一步的生物负荷降低步骤之前,应进行额外的生物负荷测试。保持时间应充分说明。
表3 接触药品或药品成分的过滤器质量文档中提供的过滤器数据
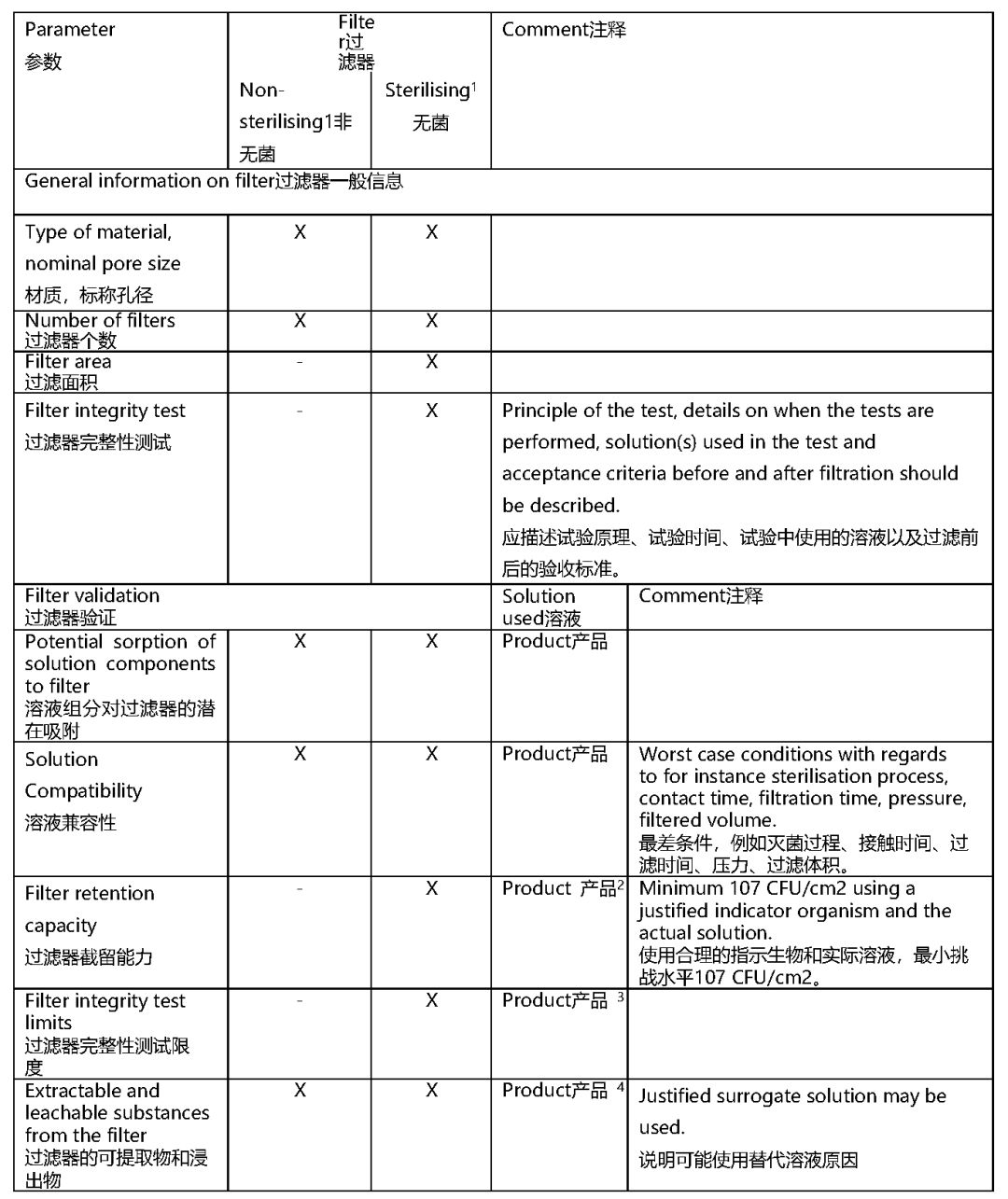
1 如《药品生产质量管理规范》附件1所定义
2 过滤器的截留能力的验证可能可以与溶液兼容性相结合。如果产品溶液对指示生物有杀菌作用,应在添加指示生物前中和。为了进行验证,应使用代表过滤器最差情况挑战的合适挑战微生物。
3 如果使用于常规生产中不同的溶液(例如注射用水)进行试验,则应在该溶液中确定限值。
4 只有当可提取物数据表明有毒成分可能迁移进入待过滤溶液时,浸出物数据有意义。
灭菌工艺选择决策树
• 虽然通过加热灭菌和通过电离辐射灭菌提供了相同的无菌保证,但通过加热灭菌具有较低的风险(例如辐射分解杂质),并且比通过电离辐射灭菌更容易控制。因此,在决策树中,热优先于电离辐射。
图1 溶液剂型产品灭菌方法选择的决策树
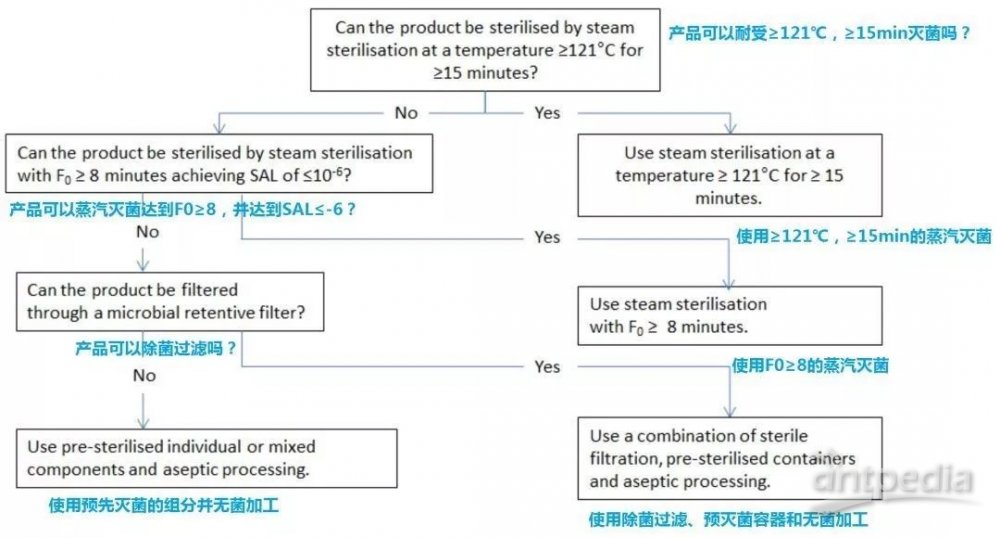
图2 非溶液剂型、半固体或干粉产品灭菌方法选择的决策树

图3 容器/包材灭菌工艺选择的决策树

请转发至朋友圈并将截图发给小编,获取该中英文完整指南译文,谢谢。
更多产品信息,请咨询默克当地销售或技术服务专员。
关于默克工艺解决方案
默克工艺解决方案是默克生命科学三大事业部之一,致力于为生物制药、化学制药企业提供产品开发、商业化生产所需全系列工具,已成为预过滤、无菌过滤、除病毒过滤、超滤、层析纯化、一次性生产、培养基、生物反应器、缓冲液、药用原辅料、工程技术及验证领域的全球领导者。默克工艺解决方案的成功源于对高质量产品、先进监管技术的不懈追求以及致力于帮助客户实现其需求的精神。

04-26 赛默飞生命科学
【反射颜色测量】典型配置、硬件说明、软件操作04-26
【文末福利】东北人做实验,爆笑来袭!小嘴儿叭叭的~04-25 黑龙江办事处
直播预告 | 大规模设备更新-赛默飞生命科学专场直播04-25 赛默飞生命科学
【转载】「最佳的抗皱塑形运动方案」研究表明:这两类运动可有效延缓皮肤衰老,特别是对于女性!04-25 生物谷
【有奖征集】「寻找实验室最美身影」主题摄影大赛火热征稿中04-25
让你的干细胞研究“研”值飙升的秘密04-25 赛默飞生命科学
CIBF2024|H.E.L诚邀新老朋友莅临指导04-25 Don Lin
苏州佳谱科技有限公司参与制定国家标准,助力水泥窑固体废物处理技术规范发展04-25
DW行业解决方案|食品安全微生物实验室能力建设04-24 DW
全国排名公布!色谱大赛战况激烈,高手如云!04-24 市场宣传部
在众多可能中,找到你的“那一个”04-24 赛默飞生命科学
融合创新,质领未来—钢研纳克蝉联检测及科学仪器行业重磅奖项04-24 钢研纳克
4月24日直播 | 使用安捷伦Seahorse技术快速精准检测线粒体毒性04-24 安捷伦细胞分析
Nature 子刊|陆军军医大肿瘤微环境新成果,一作分享研究思路04-24 转载自生物学霸
4月25日直播 | 中国PIC/S成员资格对监管实验室的影响与含义04-24 安捷伦细胞分析
它来了,符合国家卫生行业标准的流式细胞仪性能校准服务!04-24 安捷伦科技
剑桥大学/阿斯利康利用RTCA揭示cGAS-STING 该明星分子在神经小胶质细胞炎症中的机制04-24 安捷伦细胞分析
4月26日直播 | 类器官研究大讲堂 开启精准医学新时代04-24 安捷伦细胞分析
追忆星空回响,坚守使命信念——中科科仪与“东方红一号”的峥嵘岁月04-24