超干货 | 万字长文,揭秘scATAC-seq分析全过程!
2022-08-30 01:32:32, 欧易生物 上海欧易生物医学科技有限公司

Signac是一个用于分析单细胞染色质数据的综合工具包,由美国纽约基因组中心Rahul Satija和Tim Stuart联合开发,并于2021年11月1日发表于《Nature Methods》(IF:28.55)上[1]。Signac支持对单细胞染色质数据进行末端到末端分析,包括峰值检测、量化、质量控制、降维、聚类、与单细胞基因表达数据集的集成、DNA基序分析和交互式可视化等。而且Signac还能够与Seurat,MACS2,chromVAR等软件无缝衔接,促进了多种多模式单细胞染色质数据的分析,比如染色质可及性与基因表达、蛋白质丰度和线粒体基因型的共同检测等。目前Signic已经更新至1.7.0版本,可以参考官网安装链接进行安装。
今天,我们将以10x Genomics提供的人类外周血细胞数据作为演示数据进行分析,分析内容涉及数据获取、数据质控、降维聚类、基因活性矩阵创建、TF motif 矩阵创建、与scRNA-seq数据整合以及差异peaks与差异motif查找的全过程,希望对大家有所帮助。
获取数据
首先从官网获取我们接下来需要用到的分析数据,主要包括cellranger质控后的peak/cell 矩阵文件(h5文件),metadata文件(样本以及数据相关信息文件,.csv格式),fragments文件及其索引文件(tsv.gz和tbi文件),可以在终端,利用wget命令进行获取,具体命令如下:
wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_singlecell.csvwget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_fragments.tsv.gzwget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_fragments.tsv.gz.tbi
然后,我们需要安装并载入相关依赖包,除了Signac包,我们还要用到Seurat, GenomeInfoDb, EnsDb.Hsapiens.v75, ggplot2, patchwork等函数包,并利用set.seed函数设置随机种子以便结果可重复。载入命令如下:
library(Signac)library(Seurat)library(GenomeInfoDb)library(EnsDb.Hsapiens.v75)library(BSgenome.Hsapiens.UCSC.hg19)library(ggplot2)library(patchwork)library(TFBSTools)library(JASPAR2020)set.seed(1234)
构建seurat对象
在对数据进行下游分析之前,我们需要将从cellranger-atac中获得的结果,转化为seurat对象。
#读取h5文件counts <- Read10X_h5(filename = "atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")#读取metadatametadata <- read.csv( file = "atac_v1_pbmc_10k_singlecell.csv", header = TRUE, row.names = 1)#创建对象chrom_assay <- CreateChromatinAssay( counts = counts, sep = c(":", "-"), genome = ''hg19'', fragments = ''atac_v1_pbmc_10k_fragments.tsv.gz'', min.cells = 10, min.features = 200)pbmc# An object of class Seurat # 87561 features across 8728 samples within 1 assay # Active assay: peaks (87561 features, 0 variable features)pbmc <- CreateSeuratObject( counts = chrom_assay, assay = "peaks", meta.data = metadata)
构建好seurat对象之后,我们需要把基因注释文件信息添加到seurat对象中,添加命令如下:
#从EnsDB中提取基因注释信息annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)#因为要使用hg19基因组,将数据设定为UCSC格式seqlevelsStyle(annotations) <- ''UCSC''#添加注释信息到对象当中Annotation(pbmc) <- annotations
构建好的seurat对象信息如下图所示,构建完毕后我们可以接着进行下面的分析。
pbmc[[''peaks'']]## ChromatinAssay data with 87561 features for 8728 cells## Variable features: 0 ## Genome: hg19 ## Annotation present: TRUE ## Motifs present: FALSE ## Fragment files: 1
质控过滤
为进一步确保结果的可靠性,保证数据质量,我们需要根据以下5个指标,再次进行过滤,指标如下:
• nucleosome_signal:单核小体与无核小体片段的大致比率,反映 DNA 开放区域的长度信息,DNA 开放区域的长度通常会在 1~2 个核小体长度(147bp)范围内,一般会过滤掉nucleosome_signal>4的barcodes。
•TSS 富集分数:以 TSS 为中心的片段与 TSS 侧翼区域中的片段的比率即为TSS富集分数(ENCODE)。TSS 富集分数较低表明TSS区域的fragments占用细胞全部fragments的比例较低,数据质量较差。我们可以使用 TSSEnrichment() 函数为每个细胞计算此指标,并将结果存储在列名 TSS.enrichment下的元数据中。
•peaks中的片段总数:衡量细胞测序深度/复杂性的指标。由于测序深度低,质控可能需要排除reads数很少的细胞。reads数极高的细胞可能代表了双细胞、细胞核团块。
•peaks中片段的比例:比例较低的细胞(即<15-20%)通常表示在质控中应删除的低质量细胞或技术误差。
•比对到基因组“blacklist”区域中的reads比率:ENCODE 数据库中收集了 blacklist 区域(基因组总反常的或者无论在二代测序的哪个实验中都是高信号的区域,排除掉这些区域可以更好的分析功能基因组数据),因此要求 barcode 中比对到 blacklist 的 fragments 的比例要低于一定的阈值。
最后三个指标结果,我们可以从cellranger结果文件中获得,下面是各指标获得方法:
#计算每个细胞的nucleosome signalpbmc <- NucleosomeSignal(object = pbmc)#计算TSS富集分数pbmc <- TSSEnrichment(object = pbmc, fast = FALSE)#计算blacklist ratio和peaks中的片段数pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100pbmc$blacklist_ratio <- pbmc$blacklist_region_fragments / pbmc$peak_region_fragments我们可以先看一下在质控前,各指标数据的分布情况,以VlnPlot函数进行展示。VlnPlot( object = pbmc, features = c(''pct_reads_in_peaks'', ''peak_region_fragments'', ''TSS.enrichment'', ''blacklist_ratio'', ''nucleosome_signal''), pt.size = 0.1, ncol = 5)
然后根据以上5个质控指标对数据进行质控,详细阈值如下所示,完成质控最终获得7060个高质量细胞。
pbmc <- subset( x = pbmc, subset = peak_region_fragments > 3000 & peak_region_fragments < 20000 & pct_reads_in_peaks > 15 & blacklist_ratio < 0.05 & nucleosome_signal < 4 & TSS.enrichment > 2)pbmc# An object of class Seurat # 87561 features across 7060 samples within 1 assay # Active assay: peaks (87561 features, 0 variable features)
对质控后的各项指标进行展示,具体情况如下:
降维聚类
在对数据完成质控后,我们需要对数据进行归一化处理,归一化处理的目的是为了纠正细胞测序深度的差异,能够给稀有峰更高的值,不同于单细胞转录组归一化的过程,Signac采用TF-IDF的方法对数据进行归一化处理。
在对数据进行降维处理前,单细胞转录组由于数据之间动态变化区间不大,往往需要挑选一些高变基因进行后续的降维,同样在scATAC中,我们可以挑选top n的peaks用于降维,或者利用FindTopFeatures()函数过滤掉部分peaks用于后续的分析。接下来我们使用上面选择的peaks在TD-IDF矩阵上进行奇异值分解(SVD),这样我们就完成了对scATAC的降维过程,和单细胞转录组的PCA降维过程不同,我们采用TF-IDF与SVD相结合的两步降维方法进行降维,这种两步降维方法也被称为潜在语义索引法(LSI)。
降维完成之后,我们采用和单细胞转录组相同的流程进行图形化展示和聚类,具体代码如下:
#降维pbmc <- RunTFIDF(pbmc)pbmc <- FindTopFeatures(pbmc, min.cutoff = ''q0'')pbmc <- RunSVD(pbmc)#聚类pbmc <- FindNeighbors(object = pbmc, reduction = ''lsi'', dims = 2:30)pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)#UMAP展示pbmc <- RunUMAP(object = pbmc, reduction = ''lsi'', dims = 2:30)#plot结果DimPlot(object = pbmc, label = TRUE)
创建基因表达活性矩阵
从UMAP图中我们可以看到,人的外周血中存在多个细胞类群,但要在scATAC-seq数据中对这些类群进行鉴定,可能存在很大的挑战。因此,我们尝试通过评估与每个基因相关的染色质可及性来量化基因组中每个基因的表达活性,构建出类似单细胞转录组的基因表达活性矩阵,以基因表达活性矩阵进行后续的细胞类型鉴定可能会更简单一些。
基因表达活性矩阵过程如下:
(1)我们从EnsembleDB中提取了人类基因组的基因坐标,并将其扩展到TSS上游2kb区域(启动子区域的可及性与基因表达相关);
(2)我们使用FeatureMatrix函数计算映射到这些区域的每个细胞的片段数;
(3)最终将返回一个稀疏矩阵,即为基因活性矩阵。该过程Signac已封装至GeneActivity函数中,直接执行下面代码即可。
gene.activities <- GeneActivity(pbmc)#将基因活性矩阵添加至seurat对象中pbmc[[''RNA'']] <- CreateAssayObject(counts = gene.activities) #进行normalizepbmc <- NormalizeData( object = pbmc, assay = ''RNA'', normalization.method = ''LogNormalize'', scale.factor = median(pbmc$nCount_RNA) )
基因活性矩阵创建完成之后,我们就可以绘制一些细胞类特异marker gene的表达量图,来辅助我们进行细胞类型鉴定等工作。
DefaultAssay(pbmc) <- ''RNA''FeaturePlot( object = pbmc, features = c(''MS4A1'', ''CD3D'', ''LEF1'', ''NKG7'', ''TREM1'', ''LYZ''), pt.size = 0.1, max.cutoff = ''q95'', ncol = 3)

创建motif矩阵
除了创建基因活性矩阵,我们还可以计算motif deviation score,简单来说,它是通过一定的算法原理,利用公共TF motif数据库,给每个细胞的每个TF对应的motif进行一个打分值,值的高低代表该motif在该细胞的调控活跃程度[4]。Signac是借助了chromVAR函数实现了该功能。
首先,我们需要在seurat对象中添加motif相关的信息,代码如下:
DefaultAssay(pbmc)<-"peaks"#获取数据库motif信息pfm <- getMatrixSet( x = JASPAR2020, opts = list(collection = "CORE", tax_group = ''vertebrates'', all_versions = FALSE))#创建motif矩阵pbmc <- AddMotifs( object = pbmc, genome = BSgenome.Hsapiens.UCSC.hg19, pfm = pfm)添加motif信息后,我们开始计算motif deviation score,代码如下:pbmc <- RunChromVAR( object = pbmc, genome = BSgenome.Hsapiens.UCSC.hg19)
我们可以利用MotifPlot函数和FeaturePlot函数对部分motif进行可视化。
MotifPlot( object = pbmc, motifs = c("MA0497.1","MA1151.1","MA0773.1"))

说明:
“MA0497.1","MA1151.1","MA0773.1”为motif ID,其对应的motif name即TF 为: "MEF2C", "RORC" ,"MEF2D",MotifPlot默认以TF作为图的标题。
FeaturePlot( object = pbmc, features = c("MA0497.1","MA1151.1","MA0773.1"), min.cutoff = ''q10'', max.cutoff = ''q90'', pt.size = 0.1, ncol = 3)
与scRNA-seq数据整合分析
另外,为了进一步解释scATAC-seq数据,我们可以利用来自同一生物组织的scRNA-seq数据对细胞进行分类。scATAC-seq数据与scRNA-seq数据的整合主要是基于scATAC-seq中的基因活性矩阵与scRNA-seq基因活性表达矩阵的相关性,然后根据scRNA中对细胞类型的定义转移至对应的scATAC-seq数据中[2],本文下载了人类 PBMC 的预处理 scRNA-seq 数据集,同样由10x官方提供.
预处理后数据的下载地址为:
https://signac-objects.s3.amazonaws.com/pbmc_10k_v3.rds
具体分析代码如下:
DefaultAssay(pbmc)<-"RNA"pbmc_rna <- readRDS("pbmc_10k_v3.rds")#创建两组学间的anchorstransfer.anchors <- FindTransferAnchors( reference = pbmc_rna, query = pbmc, reduction = ''cca'')#标签转移predicted.labels <- TransferData( anchorset = transfer.anchors, refdata = pbmc_rna$celltype, weight.reduction = pbmc[[''lsi'']], dims = 2:30)pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
将预测的细胞类型标签添加至pbmc的metadata当中后,我们可以分别绘制基于scATAC-seq和scRNA-seq数据的umap图。
#scRNA细胞类型umapplot_rna<- DimPlot( object = pbmc_rna, group.by = ''celltype'', label = TRUE, repel = TRUE) + NoLegend() + ggtitle(''scRNA-seq'')#scATAC细胞类型umapplot_atac <- DimPlot( object = pbmc, group.by = ''predicted.id'', label = TRUE, repel = TRUE) + NoLegend() + ggtitle(''scATAC-seq'')plot_rna+plot_atac
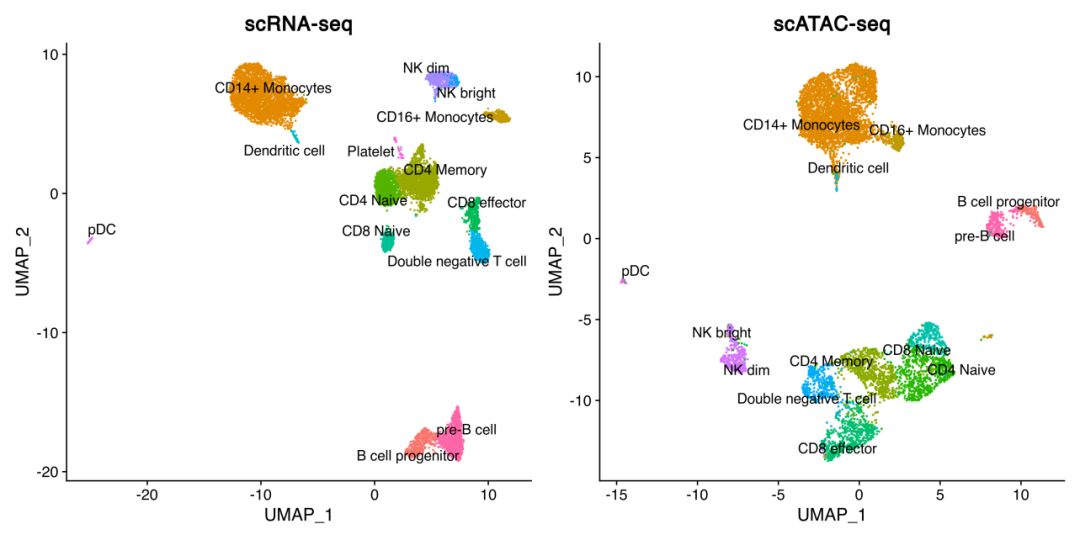
对于scATAC数据,我们可以基于预测的细胞类型,结合降维所得的簇,重新对每个簇进行细胞类型注释,注释代码及结果如下:
pbmc <- subset(pbmc, idents = 14, invert = TRUE)pbmc <- RenameIdents( object = pbmc, ''0'' = ''CD14 Mono'', ''1'' = ''CD4 Memory'', ''2'' = ''CD8 Effector'', ''3'' = ''CD4 Naive'', ''4'' = ''CD14 Mono'', ''5'' = ''DN T'', ''6'' = ''CD8 Naive'', ''7'' = ''NK CD56Dim'', ''8'' = ''pre-B'', ''9'' = ''CD16 Mono'', ''10'' = ''pro-B'', ''11'' = ''DC'', ''12'' = ''NK CD56bright'', ''13'' = ''pDC'')pbmc$clusters<-Idents(pbmc)plot_annotion<-DimPlot( object = pbmc, group.by = ''clusters'', label = TRUE, repel = TRUE)+ NoLegend()+ggtitle(''Annotion'')plot_predict<-DimPlot( object = pbmc, group.by = ''predicted.id'', label = TRUE, repel = TRUE) + NoLegend() + ggtitle(''Predicted'')plot_annotion+plot_predict
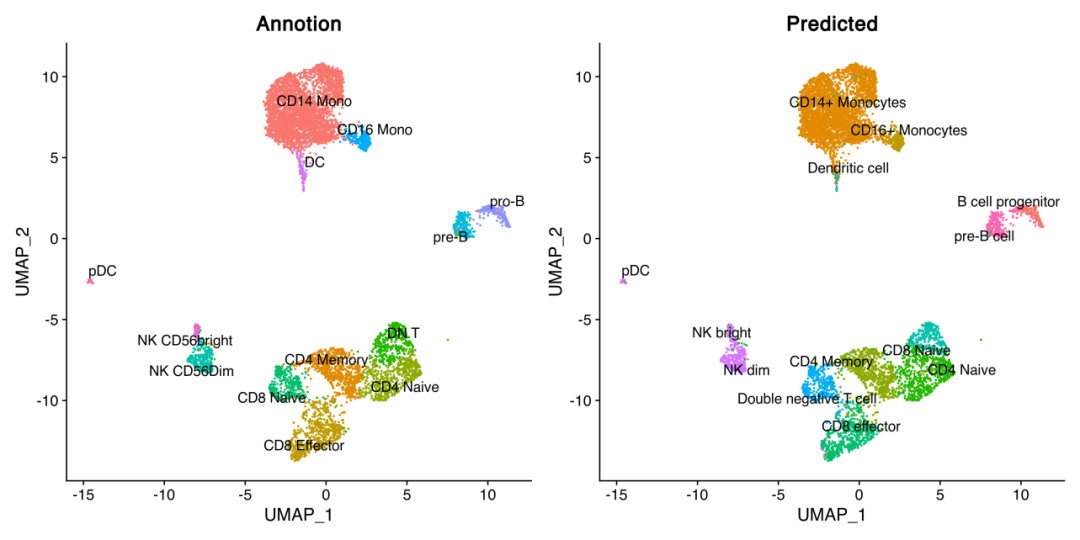
从umap图中我们可以大致看出,预测和实际降维聚类结果基本一致。
查找差异peaks及差异motif
在完成细胞类型鉴定之后,我们可以寻找不同细胞类型之间差异的peak以及差异的motif。不同于单细胞转录组,在对差异peaks和motif的寻找中,我们采用是逻辑回归( LR )的方法[3],并将片段总数添加在潜在变量中,主要是为了减少测序深度对结果的影响。另外,由于scATAC数据的稀疏性,我们通常会把min.pct值降低到0.05,以"CD4 Naive"和"CD14 Mono"细胞为例,具体的代码及结果如下:
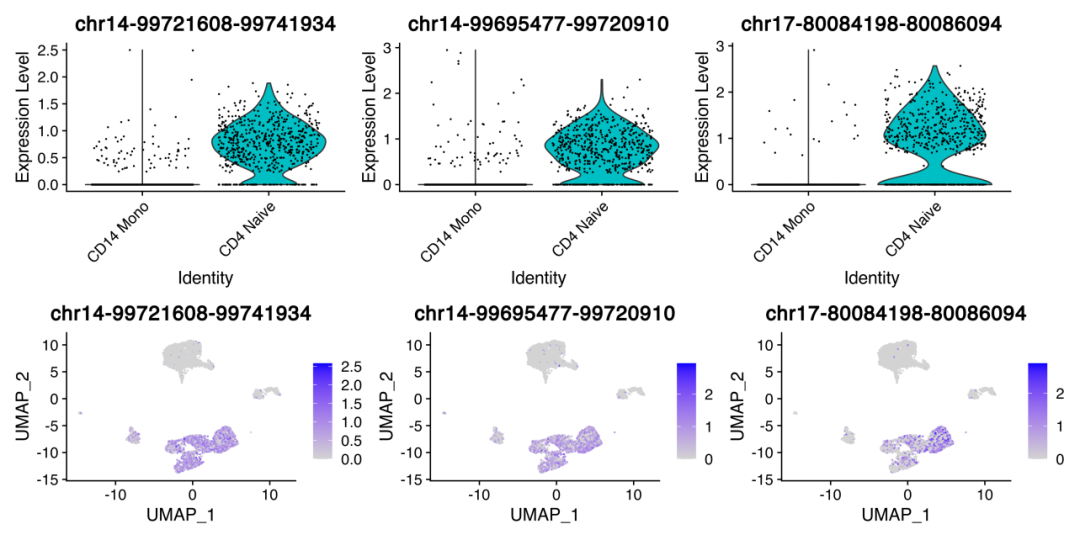
#查找差异peaksDefaultAssay(pbmc) <- ''peaks''da_peaks <- FindMarkers( object = pbmc, ident.1 = "CD4 Naive", ident.2 = "CD14 Mono", min.pct = 0.05, test.use = ''LR'', latent.vars = ''peak_region_fragments'')head(da_peaks)# p_val avg_log2FC pct.1 pct.2 p_val_adj# chr14-99721608-99741934 1.622701e-307 0.7746012 0.879 0.023 1.420853e-302# chr14-99695477-99720910 3.713144e-241 0.7611656 0.825 0.023 3.251266e-236# chr17-80084198-80086094 1.789358e-231 0.9220662 0.695 0.006 1.566780e-226# chr7-142501666-142511108 1.284659e-230 0.6751046 0.767 0.032 1.124860e-225# chr2-113581628-113594911 2.820671e-212 -0.7572235 0.055 0.693 2.469808e-207# chr6-44025105-44028184 3.989239e-206 -0.7447979 0.063 0.654 3.493017e-201
我们利用VlnPlot和FeaturePlot对数据进行可视化展示一下。
plot1<-VlnPlot( object = pbmc, features = rownames(da_peaks)[1:3], pt.size = 0.1, idents = c("CD4 Naive","CD14 Mono"))plot2<-FeaturePlot( object = pbmc, features = rownames(da_peaks)[1:3], pt.size = 0.1, ncol=3)plot1/plot2
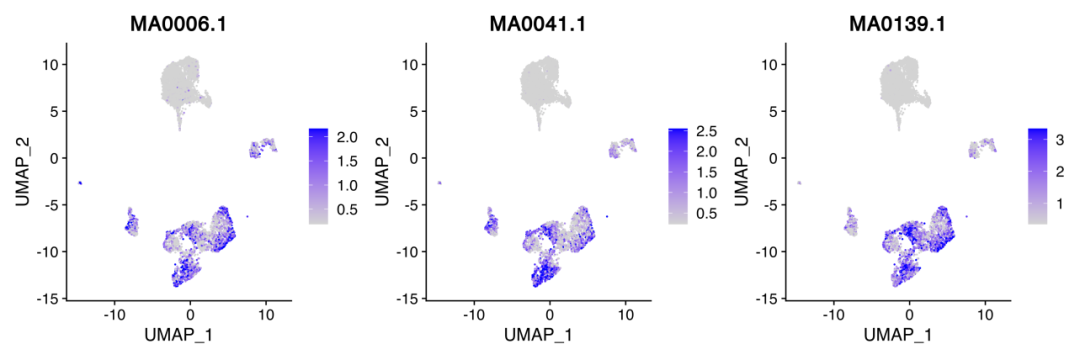
采用同样的方式,我们来寻找这两个细胞类型之间差异的motif。
DefaultAssay(pbmc) <- ''chromvar''da_motif <- FindMarkers( object = pbmc, ident.1 = "CD4 Naive", ident.2 = "CD14 Mono", min.pct = 0.05, test.use = ''LR'', latent.vars = ''peak_region_fragments'')head(da_motif)# p_val avg_log2FC pct.1 pct.2 p_val_adj# MA0006.1 0 3.520682 0.859 0.067 0# MA0041.1 0 3.678268 0.838 0.030 0# MA0139.1 0 6.070886 0.942 0.021 0# MA0151.1 0 3.799408 0.915 0.022 0# MA0476.1 0 -9.831908 0.007 0.945 0# MA0478.1 0 -10.893881 0.009 0.949 0FeaturePlot( object = pbmc, features = rownames(da_motif)[1:3], min.cutoff = ''q10'', max.cutoff = ''q90'', pt.size = 0.1, ncol=3)
今天,我们关于scATAC-seq数据的基础分析介绍就到这里了,后面我们将就scATAC-seq数据的深度挖掘和解析进一步介绍,欢迎关注。
参考文献
1.Stuart, T., Srivastava, A., Madad, S. et al. Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341 (2021). https://doi.org/10.1038/s41592-021-01282-5
2.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888-1902.e21. doi:10.1016/j.cell.2019.05.031
3.Ntranos, V., Yi, L., Melsted, P. et al. A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat Methods 16, 163–166 (2019). https://doi.org/10.1038/s41592-018-0303-9
4.Cusanovich DA, Daza R, Adey A, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910-914. doi:10.1126/science.aab1601
- OE BIOTECH
OE BIOTECH -
猜你想看
1、Front Endocrinol | FFPE样本 VS 新鲜样本,全转录组测序的肾上腺皮质肿瘤RNA表达高度相似?
2、项目文章 l 四川农业大学研究团队发现转录因子ZmTCP7对玉米淀粉合成积累的调控机制
3、四篇植物单细胞高分文章:教你玩转植物单细胞发文新思路!
4、Monocle3单细胞拟时序轨迹方法新升级,还不快来试试?
07-01 英斯特朗
连载 | 药物一致性评价与粒度分析(三)07-01 欧美克仪器
【仪器百科】LS-909丨干湿二合一激光粒度分析仪07-01 欧美克仪器
标准物质解决方案 | PFASs(全氟及多氟化合物)06-29
第九期阿尔塔有约 | 环境专题【新污染物:PFAS】技术研讨会精彩回顾及提问解答06-29
“绿色技术范式”,分析化学未来发展方向——访中国分析测试协会副理事长、辽宁省分析科学研究院原院长刘成雁教授06-29 转载仪器信息网
华西医院-标准型数显脑立体定位仪、双通道体温维持仪、体式显微镜安装完成06-29 迈越生物
科鉴检测助力2家仪器企业获得首批产品可靠性认证证书06-28 科鉴检测
德国耶拿:锂电池生命周期分析解决方案06-28 德国耶拿
AI已来!生命科学本科教学如何紧跟技术浪潮06-28 Opentrons
盛瀚售后,五星级服务的秘诀是什么?06-28 SHINE
专为汽车制造商打造的柔性解决方案——实现制程控制06-28
西北工业大学-脑立体定位仪安装完成06-28 迈越生物
会议邀请 | 第九届海上检验医师论坛06-28
卓立要闻 | 创新发展ing…6月卓立“大事小情”速览06-28 光电行业都会关注
打造信任合作伙伴!2024年度卓立汉光客户满意度调查开启06-28 光电行业都会关注
如何挑选适用于三阶光学非线性的测量系统?Z扫描测量系统来助力!06-28 光电行业都会关注
招聘启事—中国科学院沈阳自动化研究所微纳光学测量表征技术课题组06-28 光电行业都会关注
谱育科技作为主要完成方 荣获2023年度国家科学技术进步一等奖和二等奖06-28 点击关注→
仪器原理丨顶空仪与吹扫捕集仪科普小知识06-28 天美色谱