Monocle3单细胞拟时序轨迹方法新升级,还不快来试试?
2022-08-20 06:47:20, 欧易生物 上海欧易生物医学科技有限公司

Monocle3背景简述

在外界刺激或者发育过程中,不同细胞的功能会发生不同的变化,从而导致细胞的表达量呈现出差异性。根据每个细胞的基因表达情况对这些细胞进行排序,能够进一步推断出细胞发育的轨迹。
Junyue Cao(曹俊越)等[1]在进行小鼠的发育生物学研究时,由于数据缺少一些祖先的状态,且已知的一些细胞类型来自多个转录不同的谱系,因此为了克服这些限制,同时将拟时序方法扩展到数百万个细胞,于是开发了以PAGA算法[2]为蓝本的新方法,即Monocle3。该方法因其高效的运算速度和美观的可视化,迅速登上了单细胞拟时序分析的舞台,并已在《Nature》杂志上发表。
Monocle3软件介绍
相比以往熟知的Monocle2,Monocle 3经过了重新设计,使之可以用于分析大型、复杂的单细胞数据集。Monocle 3的核心算法具有高度可扩展性,可以处理数百万个细胞。软件主要可以执行三类分析:1)对细胞聚类、分群和计数;2)构建单细胞轨迹;3)差异表达分析。Monocle 3在R语言(v4.1.0或更高版本)环境中运行,同时需要R包Bioconductor(v3.14或更高版本)和monocle3(v1.2.7或更高版本)来访问最新功能。
Monocle3原理介绍
1. 使用UMAP降维
统一流形逼近和投影 (UMAP) 是一种降维技术,Monocle3首先将数据投影到一个低维空间,基于黎曼几何和代数拓扑的算法来执行降维和数据可视化。与t-SNE相比,UMAP的可视化更具有竞争力,最显著的是提高了运行速度并更好地保存了数据的全局结构。结果发现,UMAP的计算效率显著地加快了对小鼠胚胎数据的分析。UMAP在大约3个CPU小时内完成了处理200万个单元格的数据集,而t-SNE花费了超过64个CPU小时。当然,Monocle3也可以直接导入Seurat的降维结果进行分析。
2. 将细胞划分为不连续的轨迹
细胞的分化总是具有连续性和离散性的。当数据量很大时,Monocle2可能会错误地把本来不在同一个轨迹上的细胞认为是在同一个轨迹上,而PAGA(基于分区的图抽象方法,Partition-based graph abstraction)方法则解决了这个困难。
3. 轨迹图学习
Monocle3在相同的低维空间进行轨迹图学习,来表示细胞在发育过程中可能采取的路径。对此,Monocle3使用了一个基于SimplePPT算法的图嵌入过程。另外,Monocle3还有几点升级:1)直接在UMAP空间(默认为三维的)中绘制轨迹主图,避免直接处理成千上万个单细胞数据;2)能够平滑和细化主图以排除小分支,从而消除噪声分支;3)适用于环路轨迹,而不要求分化轨迹一定是树形结构。
4. 计算伪时间
为了计算细胞级别的伪时间,Monocle3开发了一种适用于具有数百万细胞数据集的投影策略。该策略的原理是,以主图为指导,在所有单元上构造一个图ψ,然后通过计算每个细胞与选择的一个根或多个根的距离作为伪时间。Monocle3是一个半监督的拟时序分析,因此需要通过生物学背景来选择合适的起始点。
5. 识别表达基因
为了识别在发育轨迹中表达变化不同的基因,Monocle3运用了一种常用于分析空间数据的统计检验。Moran’s I统计量(莫兰指数)是一个多向和多维空间自相关的度量。该统计量通过最近邻图编码数据点之间的空间关系,使其特别适合于分析大型单细胞转录组数据集。Moran指数的范围在-1~1之间,0代表此基因没有空间共表达效应,1代表此基因在空间距离相近的细胞中表达值高度相似,小于0的Moran指数一般都没有统计学意义。
Monocle3分析步骤
1. 数据导入
首先将Seurat对象转变成软件可以识别的cds对象。将Seurat对象拆分成三个模块:基因的表达矩阵(稀疏矩阵)、metadata细胞属性信息、基因属性信息。
生成对应的三个模块数据:
data <- GetAssayData(M_3h, assay = ''RNA'', slot = ''counts'') #细胞的表达矩阵new_pd = M_3h@meta.data #meta信息fData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data)) #基因属性信息
使用三个模块数据创建cds对象:
cds <- new_cell_data_set(expression_data=data,cell_metadata=new_pd,gene_metadata= fData)2. 数据预处理
数据预处理包括对数据进行归一化、标准化、去除批次、PCA降维等,并设置一个PCA维度(这里设置为10),可以用plot_pc_variance_explained()函数来展示维度。
cds <- preprocess_cds(cds, num_dim = 10)3. 非线性降维和可视化
一般使用UMAP或t-SNE进行非线性降维和可视化。Moncle3更推荐使用UMAP方法,处理单细胞数据更快。
cds <- reduce_dimension(cds,preprocess_method = ''PCA'',reduction_method = ''UMAP'')4. 聚类分析
聚类分析可以判别哪些细胞是来自一个祖先(一个发育轨迹上)。
cds <- cluster_cells(cds)5. 拟时序分析
拟时序分析用来找出每个群内的细胞发育轨迹。利用拟时序分析的结果可以绘制拟时序图。
cds <- learn_graph(cds) #轨迹推断plot_cells(cds, color_cells_by = "partition", cell_size = 0.5)
6. 选择合适的起点
可以通过算法计算起始点,也可以通过手动选择起始点,考虑到生物学意义,推荐手动设置起点。
cds <- order_cells(cds, root_cells = root.cell) #选择对应的细胞群作为起点7. 下游分析
下游分析包括差异基因分析、单个基因分析(判断基因的激活顺序)等。
Monocle3结果展示

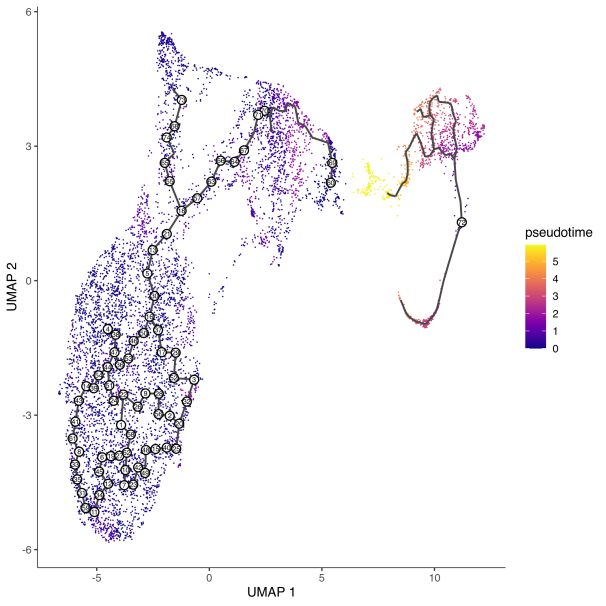
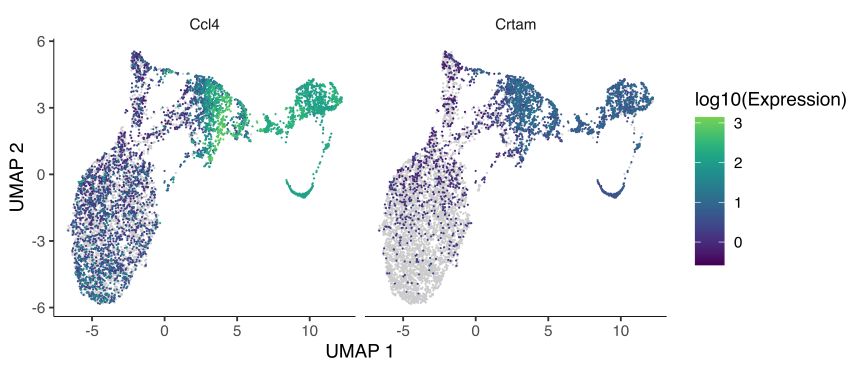

今天我们对Monocle3的简介就到此,不知道您有没有get到这款好用便捷又快速的软件呢?有需要的老师赶快试试吧!
参考文献:
[1] Cao, J., Spielmann, M., Qiu, X. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496-502 (2019).
https://doi.org/10.1038/s41586-019-0969-x
[2] Wolf, F.A., Hamey, F.K., Plass, M. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 20, 59(2019).
https://doi.org/10.1186/s13059-019-1663-x
猜你想看
1、Plant Physiol l 西北农林科技大学李强王保通研究团队发现谷胱甘肽S-转移酶增强小麦抗白粉病的分子机制
2、Cell Discov | 单细胞转录组测序助力解析圆锥角膜的发病机制

END
排版人:小久
原创声明:本文由欧易生物(OEBIOTECH)学术团队报道,本文著作权归文章作者所有。欢迎个人转发及分享,未经作者的允许禁止转载。


点击“阅读全文” 收获更多精彩
04-23 光电行业都会关注
展会回顾|“融两业共生之力 筑湾区超级枢纽”2026大湾区创新生态大会04-22 谱临晟
荧飒光学践行企业社会责任,赋能光电人才高质量培养04-22 荧飒光学
报名通知丨英斯特朗塑料力学测试高阶培训研讨会04-22 英斯特朗
成都科林分析邀您共赴TFF·2026酒类风味分析与感官评价暨创新技术论坛,期待与您相遇!04-22
吉艾姆4月双展齐发 | 武汉科仪展+脂在浙里研讨会04-22
应用笔记 | 基于Flex自动化平台的多体液胞外囊泡分离及EV蛋白质组学分析流程04-21 肖伟弟 曾嘉明
CCMT2026开展即高能 | Equator-X™ 双模式测量仪引爆全场04-21
告别预测偏差!Percepta自建专属训练库,pKa预测更准更快04-21 ACDLabs 李丹
世界地球日,查看地球的【愿望清单】04-21
【前沿激荡,智汇北京】IGC 2026圆满落幕,益世科生物共绘细胞基因治疗新蓝图04-21
硬核方案护航核安全|衡昇质谱斩获核材料检测装备大奖04-21
会议预告 | 英盛生物邀您共赴2026第六届北京临床质谱论坛!04-21
Turbiscan在陶瓷3D打印粘结剂分散稳定性表征中的应用及关键意义04-21 大昌华嘉
GranuCharge (粉体静电分析仪)用于研究湿度对粉末表面性能的影响04-21 大昌华嘉
生物打印鼻软骨的理想材料:GelMA水凝胶的力学与细胞外基质平衡新探索04-21 大昌华嘉生命科学
Biolin Theta系列接触角测量 | 如何在表面表征应用中使用接触角:前进角04-21 大昌华嘉
大昌华嘉科学仪器荣获Novasina 2025年度“成长与创新先锋奖”!04-21 大昌华嘉
会议预告|华大吉比爱邀您共赴第六届北京临床质谱论坛04-21 华大吉比爱
叮咚!您有一份来自地球的“绿色盲盒”待拆封04-21 安捷伦科技