“pAE会告诉你答案”——结构预测评估指标的演进(下)
2025-09-29 04:08:46, PKUMDL编辑部 TargetMol中国
本篇中最复杂的pLDDT已经介绍完了,下面就一路畅通了(希望是)。
4.和pLDDT同出一门的pTM和pAE
在AF2文章中,随pLDDT一起提出的还有pAE和pTM。
介绍pAE,就得先介绍对齐误差(aligned error,AE)。AF2论文中对对齐误差的定义是模型中残基 i 的 N、Cα 和 C 原子与实验结构中同一残基 i 的 N、Cα 和 C 原子进行叠合后,模型中残基 j 的 Cα 原子与实验结构中残基 j 的 Cα 原子之间的距离。而pAE就是对对齐误差的预测。
和pLDDT类似,pAE也是通过交叉熵作为损失函数进行训练的。不同的是,pAE接受的输入是pair representation,然后将误差离散化到64bins的区间。之后同样以pAE和实际对齐误差的交叉熵作为模型训练的损失函数,实现pAE预测头对预测结构中每个残基对的对齐误差的预测。推理中同样也是用pAE预测头读取表示后输出特定残基对的预测对齐误差的概率分布,然后取期望得到该残基对的pAE,即
,表示第 个区间的中心值(0.25 Å、0.75 Å、……、31.75 Å); 表示残基对 落在第 个区间的概率; 。
虽然AF2的论文中没有将pAE作为一个专有名词进行介绍,但在AF2 database网站的展示结果中,pAE却是作为和pLDDT一样重要的置信度指标展示给用户参考。
除了关注局部预测误差的pLDDT,残基对叠合误差的pAE,AF2论文中还提出了关注整体叠合可信度的pTM。pTM参照的是TM-score。原始的TM-score是衡量蛋白整体结构拓扑相似度的常用指标。我到现在也没有建立起对这个指标的感性认识,所以直接把它的公式贴在这里(左右滑动查看完整公式):
其中,表示在给定叠合下模型中残基 的 Cα 原子位置与实验结构中残基 的Cα 原子位置之间的距离。 是一个缩放因子,用来降低或消除 TM 分数在无关蛋白质比对时的长度依赖性,其拟合公式为:
如果 个残基,则
而pTM则是将模型输出的预测对齐误差代入原始的TM-score计算公式,从而得到 pTM 计算公式:
即对于残基i,通过链上所有其他残基相对它的pAE累积取平均计算出它的,然后链上所有残基的最大值作为整条链的pTM。pTM和pAE训练时接受的输入及分数的离散化处理基本一样,这里不再赘述。
到此,题目中的两大指标就介绍完了。
值得一提的是,我在没有开始研究本文提到的这些指标时曾盲猜pLDDT和pAE的计算原理,当时以为pLDDT是将模型的多次预测结果进行比对,实现不依赖参考结构的置信度评估。pAE也是多次预测然后比对,确定对齐误差的大小。当时还觉得非常合理,现在看来,完全没猜对。pLDDT和pAE实际上还是在和真实结构比对的过程中学会从推理过程直接评估结构预测的置信度。这提示我们AF2输出的置信度还是依赖训练集中已存在的类似的结构模式。这也使得可能存在两类预测不准的情况。第一类是给出了准确的结构,但由于和训练集中类似结构有较大差异,此时输出的pLDDT对预测结果可能存在低估;或反之,给出了不够准确的结构,但出于和训练集中类似结构的高相似性,此时输出的pLDDT对预测结果可能存在高估。同样,pAE的估计也可能存在类似的高估或低估。这提示我们后续的ipAE讨论中,pAE指标可能存在固有的局限性。(一个开放性的问题留给读者:pAE矩阵为什么不完全对称?)
5.口口相传的ipAE和认真玩梗的ipSAE
上面介绍的指标仅到AF2,还没有涉及我最关心的PPI的置信度预测。不过经历了上面的铺垫,相信大家看到下面这些指标会觉得非常自然。
在AF2模型发布之后,Deepmind的研究团队又发布了专门针对复合物结构预测的AlphaFold-Multimer。在AlphaFold-Multimer的预印本中,他们定义了界面版本的pTM,即ipTM(interface predicted TM)【4】。和pTM对同一条链上的残基对齐误差计算TM不同,ipTM特意对链间残基的对齐误差计算TM。
因此,ipTM的计算公式和上面展示过的pTM计算公式完全相同,只是要求残基i和j来自不同链。然后他们对ipTM和蛋白质对接领域常用的复合物质量评分DockQ进行了比较,认为取得了较好的一致性。
但在ipTM提出之后,不同的研究组均指出ipTM分数受到输入序列长度的影响,即使预测出的界面相同,由于输入序列长度的差异也会产生不同的ipTM分数,同时非真实结合片段和真实结合片段可能得到一样高的ipTM分数【5】。这说明ipTM作为互作筛选打分可能不够有效。与此同时,也有研究人员提出和ipTM相同思路pDockQ作为互作的预测指标【6】。
上述这些研究主要关注已知复合物的结构预测或天然蛋白间的互作预测,对于从头设计蛋白(binder)最有效的互作评分则是由一篇2023年的nature communication文章首次明确提出【7】。那么这个分数是什么呢?答案是pAE_interaction。
这个pAE_interaction的计算非常简单,就是将AF2输出的PAE矩阵取链间部分,求平均。由于PAE矩阵中涉及链间的有两部分,所以会得到以binder链所有残基(行)和target链所有残基(列)之间的pAE和以binder残基(列)和target残基(行)之间的pAE。对这两个pAE再求平均,就得到作者所说的 pAE_interaction。作者通过实验和对以往数据的分析,说明这个分数对互作的预测已经能够取得很好的效果。
其实,在这篇文章提出之前,应该也有研究人员已经注意到了ipAE在互作预测中相对ipTM的优势。因此,有人进一步细化了ipAE的计算形式,提出了actifpTM【9】,ipSAE【10】等。
在近日的一篇预印本中,有研究者对这些相关指标进行了比较,希望确定最佳的预测指标。他们的结论是,ipSAE具有最佳的预测效果【11】。因此,我们介绍ipSAE作为结尾。
ipSAE全称基于比对误差的相互作用预测得分(interaction prediction score from aligned errors)。这同时也是作者玩的一个双关梗,源自拉丁语短语 “Rēs ipsae loquuntur”,意为“事物本身会说话”。作者希望借这个双关寓意“AlphaFold 的输出评分结果足以自证其效果”。
上面曾提到,AlphaFold团队提出的ipTM基于整条链的PAE进行ipTM的计算,这会导致ipTM分数受到链长度和不存在相互作用的无序区等因素的干扰。所以,ipSAE的提出者决定只考虑挑选对齐误差较小的链间残基对进行类似ipTM的计算,同时,ipSAE的计算中还会根据链间PAE良好的残基的数目动态调整TM公式中的d0参数。因此,ipSAE的计算公式为:

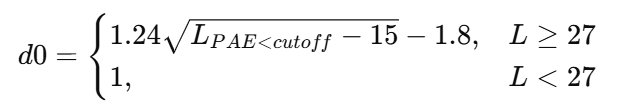
虽然上面的公式有些繁复,但简单来说就是,ipSAE只选取链间PAE较为可靠(小于给定阈值,默认为10Å)的残基以TM-score形式进行归一化,同时根据可靠残基的数量动态调整d0参数的大小以避免少数具有良好PAE的残基对对ipTM的贡献过大造成对ipTM的虚高。
对于ipSAE的非对称性质,作者选择了两个方向ipSAE中的最大值作为链间ipSAE的最终评分。由于PAE越小代表预测越准确,那么TM-score形式归一化的ipSAE则是越大代表预测越准确,作者最后选取链间ipSAE的最大值,说明作者认为只要有一侧能够取得较好的预测表现即可对预测的互作形式抱有信心。不同的是,上述对不同预测指标进行比较的预印本中的结论是,ipSAE_min能够取得比ipSAE_max更好一点的预测效果。当然,这可能与数据集有一定关系。总之,ipSAE是一个值得参考的互作评分。
——后记——
结构预测相关的评分指标还有很多,现在也不断有新的指标被提出。本文是我在试图理解pLDDT和ipSAE时向前回溯文献作的总结,所以有些不在这个演进路径中的指标就没有详细介绍(比如文中曾提到的DockQ)。也正是借助这个探究的机会,理清了之前一些错误的猜测,对这些指标的优势和局限有了更准确的认识。
感谢retreat,本文的主体是在retreat时写的,“从都市生活撤退”极大地提升了读文献和写作整理的效率,不然本文可能始终是个草稿。
希望本文对新手理解这些指标有所帮助。
全文完。
参考文献(接上篇):
[4] Evans, R., et al. "AlphaFold-Multimer Protein complex prediction with." bioRxiv (2022).
[5] Bret, Hélène, et al. "From interaction networks to interfaces, scanning intrinsically disordered regions using AlphaFold2." Nature Communications 15.1 (2024): 597.
[6] Bryant, Patrick, Gabriele Pozzati, and Arne Elofsson. "Improved prediction of protein-protein interactions using AlphaFold2." Nature Communications 13.1 (2022): 1265.
[7] Bennett, Nathaniel R., et al. "Improving de novo protein binder design with deep learning." Nature Communications 14.1 (2023): 2625.
[8]见脚本github.com/nrbennet/dl_binder_design/blob/cafa3853ac94dceb1b908c8d9e6954d71749871a/af2_initial_guess/predict.py第188-211行。
[9]Varga, Julia K., Sergey Ovchinnikov, and Ora Schueler-Furman. "actifpTM: a refined confidence metric of AlphaFold2 predictions involving flexible regions." Bioinformatics 41.3 (2025): btaf107.
[10] Dunbrack Jr, Roland L. "Rēs ipSAE loquunt: What''s wrong with AlphaFold''s ipTM score and how to fix it." bioRxiv (2025).
[11] Overath, Max Daniel, et al. "Predicting Experimental Success in De Novo Binder Design: A Meta-Analysis of 3,766 Experimentally Characterised Binders." bioRxiv (2025): 2025-08.

04-24 奥豪斯
传奇续写:奥豪斯旗下涡旋振荡器Vortex-Genie 2焕新登场04-24 奥豪斯
采用先进防静电技术,最大限度减少干扰04-24 奥豪斯
询价有礼 | 奥豪斯电化学产品解决方案04-24 奥豪斯
Angew速递:台式easyXAFS原位解析高效析氧反应的定向非晶到非晶重构04-23 Dr. Dai
五一水机关不关?测测你是水机“杀手”还是“达人”04-23 默克MilliQ纯水
Adv Sci(IF 14.1) | 上海交通大学王慧/李晓光/李辰团队通过时空蛋白质组学图谱揭示食管鳞状细胞癌早期进展的预警信号04-23
MSTD系列显微镜专用电动滑台:显微镜下图像分毫必现04-23 光电行业都会关注
分光光度计怎么用?一步步教你正确操作与数据读取方法04-23 管理员
分光光度计的工作原理详解:从朗伯-比尔定律到现代检测技术04-23 管理员
展会回顾|“融两业共生之力 筑湾区超级枢纽”2026大湾区创新生态大会04-22 谱临晟
荧飒光学践行企业社会责任,赋能光电人才高质量培养04-22 荧飒光学
天平安装丨现场直击地震对天平的影响,几十台天平瞬间“跳动”?04-22 小普
报名通知丨英斯特朗塑料力学测试高阶培训研讨会04-22 英斯特朗
成都科林分析邀您共赴TFF·2026酒类风味分析与感官评价暨创新技术论坛,期待与您相遇!04-22
吉艾姆4月双展齐发 | 武汉科仪展+脂在浙里研讨会04-22
应用笔记 | 基于Flex自动化平台的多体液胞外囊泡分离及EV蛋白质组学分析流程04-21 肖伟弟 曾嘉明
CCMT2026开展即高能 | Equator-X™ 双模式测量仪引爆全场04-21
告别预测偏差!Percepta自建专属训练库,pKa预测更准更快04-21 ACDLabs 李丹
世界地球日,查看地球的【愿望清单】04-21