Cell Metab | 巨噬细胞的代谢支持维持结肠上皮细胞稳态
2024-03-29 10:28:08, 麦特绘谱 麦特绘谱生物科技(上海)有限公司
肠道是消化和吸收食物的主要器官。肠道上皮细胞是动物机体抵御病原微生物的第一道防线, 也是黏膜机械, 免疫和化学屏障的重要组成部分, 具有吸收和屏障的双重功能。
巨噬细胞在小肠和结肠中发挥关键作用,发现结肠中的巨噬细胞与上皮隐窝细胞密切接触, 并利用雷帕霉素复合物1 (mTORC1) 信号传导途径为它们提供代谢支持。具体来说, mTORC1激活的巨噬细胞产生的多胺被上皮细胞消耗, 使其代谢重编程并促进上皮细胞的增殖,这种代谢机制在增殖性压力阶段尤为重要, 如炎症诱导的结肠炎。
2023年11月7号,奥地利维也纳医科大学 Thomas Weichhart团队在Cell Metabolism上发表题为 “Metabolic support by macrophages sustains colonic epithelial homeostasis”的研究性论文, 该研究利用代谢组学、16s rRNA测序、转录组学和基因敲除等技术, 揭示髓系巨噬细胞通过mTORC1的激活促进多胺的生产, 结肠上皮细胞则通过摄取多胺进行代谢重编程, 加速其增殖,促进结肠上皮的有效自我更新,保护肠道免受结肠炎引起的损伤。

基因敲除技术路线

1. 巨噬细胞中mTORC1的激活可以预防DSS诱导的结肠炎
分析正常小鼠的结肠样本发现, F4/80+ (巨噬细胞标记物) 巨噬细胞与上皮隐窝细胞密切接触。考虑到代谢在巨噬细胞的稳态中发挥重要作用,而mTOR信号在整合环境营养条件中发挥重要作用。基于此,构建小鼠巨噬细胞Tsc2基因 (mTORC1的负调控因子)的特异性敲除。结果显示, 巨噬细胞中条件性敲除Tsc2能够增加上皮隐窝细胞固有层中巨噬细胞的大小和数量。利用DSS诱导结肠炎后, 发现小鼠的体重减轻量, 死亡率, 促炎因子含量和肠道通透性显著低于野生型。免疫荧光染色发现, 在野生型小鼠结肠稳态过程中, mTORC1在巨噬细胞中基本不活跃;在Tsc2特异性敲除的结肠巨噬细胞中, mTORC1通路标志物pS6保持活跃;在结肠炎期间F4/80+巨噬细胞被阶段性激活。总之,mTORC1在巨噬细胞中被诱导以应对DSS结肠炎, 并保护其免受肠道损伤。

图1. 巨噬细胞中Tsc2的缺失可预防DSS诱导的结肠炎
2. 巨噬细胞通过mTORC1促进肠道上皮细胞增殖
通过HE染色和阿利新蓝染色,发现Tsc2基因敲除小鼠结肠侵蚀性和粘液破坏性相对于野生型小鼠显著改善,推测巨噬细胞中mTORC1的激活以及Tsc2基因敲除小鼠中巨噬细胞数量的增加可能会影响上皮细胞的增殖。用mTORC1抑制剂处理Tsc2基因敲除小鼠后, 发现上皮细胞的增殖活性, 巨噬细胞的数量和mTORC1的激活被显著抑制, 其隐窝长度显著增加。

图2. 巨噬细胞通过mTORC1促进上皮细胞增殖
为了独立验证巨噬细胞中Tsc2的缺失是否促进上皮细胞更新, 使用CD11c-cre小鼠特异性敲除TSC2来验证。同样,显著增加F4/80+巨噬细胞的数量和大小, 并增强了上皮细胞的增殖, 同时隐窝的长度减少。为了更加直接验证巨噬细胞促进上皮细胞增殖,用集落刺激因子1受体(CSF1R)单克隆抗体M279清除结肠巨噬细胞。发现M279能有效清除结肠中的F4/80+细胞,减少上皮细胞层中的Ki67+细胞,从而导致了隐窝的延长。总之,结肠巨噬细胞通过mTORC1促进上皮细胞增殖。

图3. 表达cd11c的巨噬细胞促进上皮细胞更新
3. 巨噬细胞通过mTOR信号通路促进多胺的产生
为了探索巨噬细胞中的mTORC1如何促进结肠上皮细胞的增殖, 对正常状态下野生型以及结肠巨噬细胞Tsc2缺失型的结肠免疫细胞浸润, 炎症因子表达, 以及巨噬细胞极化标志物进行检测, 结果发现并无显著差异。同时,肠道微生物组学测序,结果显示,同时,肠道微生物组学测序结果显示,微生物多样性无显著差异,表明巨噬细胞中的Tsc2/ mTORC1信号通路对炎症, 细胞因子和生长因子的产生无显著影响。
鉴于mTORC1是细胞代谢的关键分子, 假设mTORC1改变结肠Tsc2基因敲除小鼠巨噬细胞代谢物的生产, 从而刺激上皮细胞增殖。为验证这一假设, 对结肠样本中的氨基酸和三羧酸循环代谢物进行代谢组学检测。结果显示11种代谢物在野生型和Tsc2基因敲除小鼠之间存在显著差异。通路富集分析结果显示, 与“亚精胺和精胺生物合成” (Spd和Spm) 及相关的生物合成途径显著富集。并且, 发现Tsc2基因敲除小鼠结肠中Spd和Spm及其前体腐胺水平显著增加, 但在DSS结肠炎中未发生显著变化。另外, Tsc2基因敲除小鼠的骨髓源性巨噬细胞 (BMDMs) 在体外产生了更多的Spd和Spm。当用雷帕霉素(mTORC1 抑制剂)对mTORC1进行抑制时, Tsc2基因敲除细胞中Spd和Spm含量的增加受到抑制。
结肠巨噬细胞的转录组学分析结果表明, 在Tsc2基因敲除小鼠中,多胺调节基因受到了显著影响, 其中精氨酸酶1 (Arg1) 在结肠中含量显著升高, 但这种差异在结肠炎期间被消除。总之, 巨噬细胞中多胺的产生以Tsc2/mTORC1依赖的方式被刺激。

图4. 巨噬细胞依赖mTOCR1途径产生多胺
4. 髓系巨噬细胞多胺的产生促进肠上皮细胞增殖
为了验证巨噬细胞多胺的产生是否能促进肠上皮细胞增殖, 对多胺合成中的关键酶进行阻断, 比如ODC1抑制剂DFMO和SAM抑制剂SAM486A来处理野生型小鼠, 结果表明, 这些抑制剂阻断了肠上皮细胞的增殖, 并减少了巨噬细胞数量。构建巨噬细胞中Arg1基因的特异性敲除, 结果显示,结肠上皮细胞增殖显著减少, 隐窝长度显著延长。此外, Arg1敲除小鼠结肠中的put、Spd和Spm水平显著降低, 并且更容易被DSS诱导为结肠炎。使用外源性重组Arg1蛋白来消耗L-精氨酸的水平也显著降低了巨噬细胞数量和结肠上皮细胞的增殖能力。
为了更准确的验证骨髓来源的巨噬细胞产生的多胺能够影响肠道上皮细胞增殖, 对野生型小鼠进行辐射, 并移植Sat1-KO或对照骨髓(SAT1是多胺代谢分解代谢途径中的一种限速酶), 结果发现,接受Sat1-KO骨髓治疗的小鼠的细胞增殖增强,隐窝长度减少。总之, 巨噬细胞和骨髓细胞中多胺产生的影响结肠上皮细胞的增殖。

图5. 髓系多胺的产生可促进上皮细胞的增殖
5. 巨噬细胞通过mTORC1促进肠道上皮细胞增殖
使用荧光探针对Spd和Spm进行标记, 发现外源Spd和Spm很容易被两种结肠细胞系HCT116和Caco-2细胞摄取, 而这种摄取容易被多胺转运抑制剂二氯化苯甲酯 (BV) 阻断。使用不同浓度的Spd和Spm对两种结肠细胞系HCT116和Caco-2进行处理。结果表明, 这两种多胺在体外都能增强结肠上皮细胞的增殖。此外, Spm(而非 Spd)直接促进了两种独立的人类结肠类器官的生长。
从Tsc2基因敲除小鼠结肠中分离EpCAM+CD44结肠上皮细胞, 并进行RNA测序 (RNA-seq)。发现其差异基因主要富集在糖酵解, 氧化磷酸化, 以及DNA修复富集途径。为了验证这一结果, 使用Seahorse XFp细胞能量代谢分析仪测定野生型和Tsc2基因敲除小鼠的糖酵解和氧化磷酸化水平。结果表明, Tsc2基因敲除的结肠上皮细胞中糖酵解和氧化磷酸化水平显著增加。同样, 将野生型和Tsc2基因敲除小鼠进行DSS诱导, 并通过pH2AX染色来测量DNA损伤。结果表明, Tsc敲除小鼠的结肠上皮细胞的DNA损伤显著减少, 而Arg1敲除小鼠的结肠上皮细胞的DNA损伤增加。总之, 结肠上皮细胞吸收了由mTORC1激活的巨噬细胞产生的外部多胺从而提升自身代谢能力, 降低DSS诱导的结肠上皮细胞DNA损伤, 从而促进肠上皮细胞增殖。
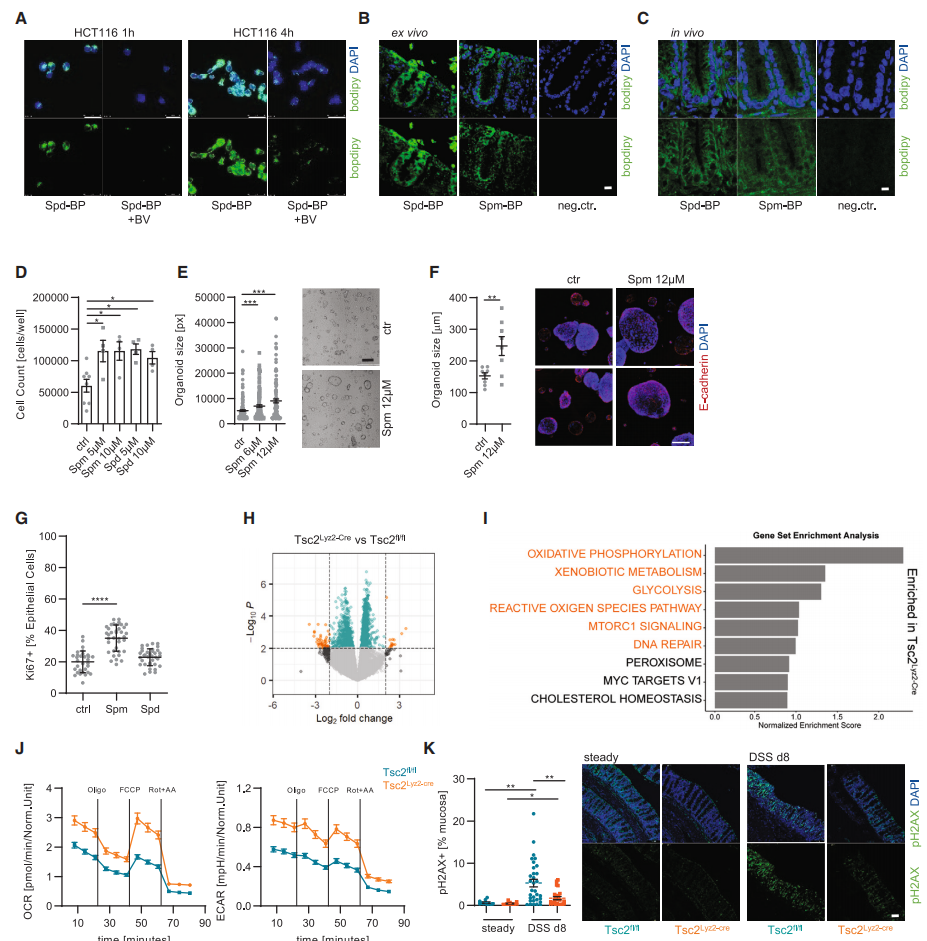
图6. 外部多胺的摄取重新连接代谢,以支持结肠上皮细胞的防御和增殖
小结
本研究发现,巨噬细胞中Tsc2的缺失激活了mTORC1信号传导,该信号传导可防止结肠炎诱导的肠道损伤,并诱导多胺亚精胺和精胺的合成。上皮细胞摄入这些多胺并重新连接其细胞代谢以优化增殖和防御。该研究结果表明,巨噬细胞充当“信使”,提供代谢支持,促进结肠上皮的有效自我更新。本研究丰富了对肠道微环境和免疫细胞在维持肠道健康中的理解,也为炎症性肠病和结直肠癌等疾病的治疗提供了新思路。

参考文献
Metabolic support by macrophages sustains colonic epithelial homeostasis. Cell Metabolism. 2023.
请扫描二维码阅读原文

绘谱帮你测
随着多胺代谢物被各大顶级学术期刊陆续报道,其延缓衰老、抗肿瘤免疫等作用日益显著,针对多胺及相关代谢通路,麦特绘谱开发了针对性靶向代谢组学产品,助力临床研究。
麦特绘谱开创性地搭建了医学领域高端代谢组学技术平台,覆盖了非靶向-全定量-代谢流等全方位的高端医学代谢组解决方案,同时全面布局微生物组学、转录组学和蛋白质组学等多组学技术服务,已成为全球多组学研究者的优选合作伙伴。麦特绘谱拥有Q1000、Q500、Q300、Q200和胆汁酸、短链脂肪酸、色氨酸及吲哚衍生物、多胺和TMAO类等各类小分子代谢物、非靶向代谢组学和同位素示踪代谢流技术等共40+系列检测方法;已为数百家三甲医院、科研院所和企业提供多组学解决方案,协助客户与合作伙伴发表SCI文章300+篇,累计影响因子3000+,平均IF>10,包括Science, Nature, Cell Metabolism ,Immunity, Gut, Hepatology, Microbiome等顶级期刊。

END
Metabo-Profile
全产品一览


项目合作成果
研究领域全集
•肠道菌群 •肿瘤/癌症 •肠道疾病 •肝脏疾病 •心血管疾病 •肥胖/糖尿病 •饮食/运动研究 •神经系统疾病 •中西药 •长寿/衰老 •免疫疾病 •铁死亡 •病毒
聚焦研究前沿
更多绘谱资源
关注了解更多

扫码关注“麦特绘谱”公众号

扫码关注“麦特绘谱”视频号

扫码关注“麦特绘谱”B站账号

扫码关注“麦特绘谱”知乎号
04-24
激光在激光粒度分析仪中的作用04-24 真理光学粒度仪
LT3600 Plus激光粒度分析仪04-24 真理光学
真理光学诚邀你参加粉体圈第八届全国氧化铝会议04-24 真理光学粒度仪
珂睿科技诚邀您参加广西分析仪器设备应用技术交流会!04-23 珂睿marketing
终于来啦,MitoTracker小包装04-23 赛默飞生命科学
践行新质生产力:Chromeleon CDS简化您的工作流04-23 飞飞
【报名火爆】2024单细胞蛋白质组学技术与产业应用研讨会线上直播通道发布!04-23
特洁安Aquafine助力河南电子半导体客户高品质生产,为“中国芯”提供有力保障04-23
隆重上市!第六代频率步进三维探地雷达突破地下调查的界限04-23 专业的
降本增效 | 奥豪斯称重产品助力制药行业04-23 奥豪斯
Aliben动态 | 俄罗斯科学院院士Valery Tuchin教授到访艾立本科技参观交流04-23 艾立本科技
春启青莲礼,血浆免费试 | 高深度血浆蛋白质组限时免费测!04-23
抢占报名 | 4月25日“御光同行”光电技术与应用研讨会昆明专场04-23 聚焦光电前沿领域
【Vocus B文献分享】一种可检测有机和无机物种的中压化学电离反应腔表征04-23
德国元素TOC总有机碳分析仪线下用户培训邀请函(第一轮通知)04-23
最高91分,机会巨大!答题赛最后12小时,通道即将关闭!04-23 市场宣传部
【 Stage-RTL反射率测试】典型配置、硬件说明、软件操作04-23
相约青岛-第六届大气臭氧污染防治研讨会04-22 TOFWERK中国
从专家共识看吉比爱如何布局质谱自动化及应对临床质谱挑战04-22 华大吉比爱