金属元素控制之法规要求
2023-04-18 01:35:59, 基泰生物 上海基泰生物科技有限公司
金属元素控制之法规要求
一、元素杂质分类
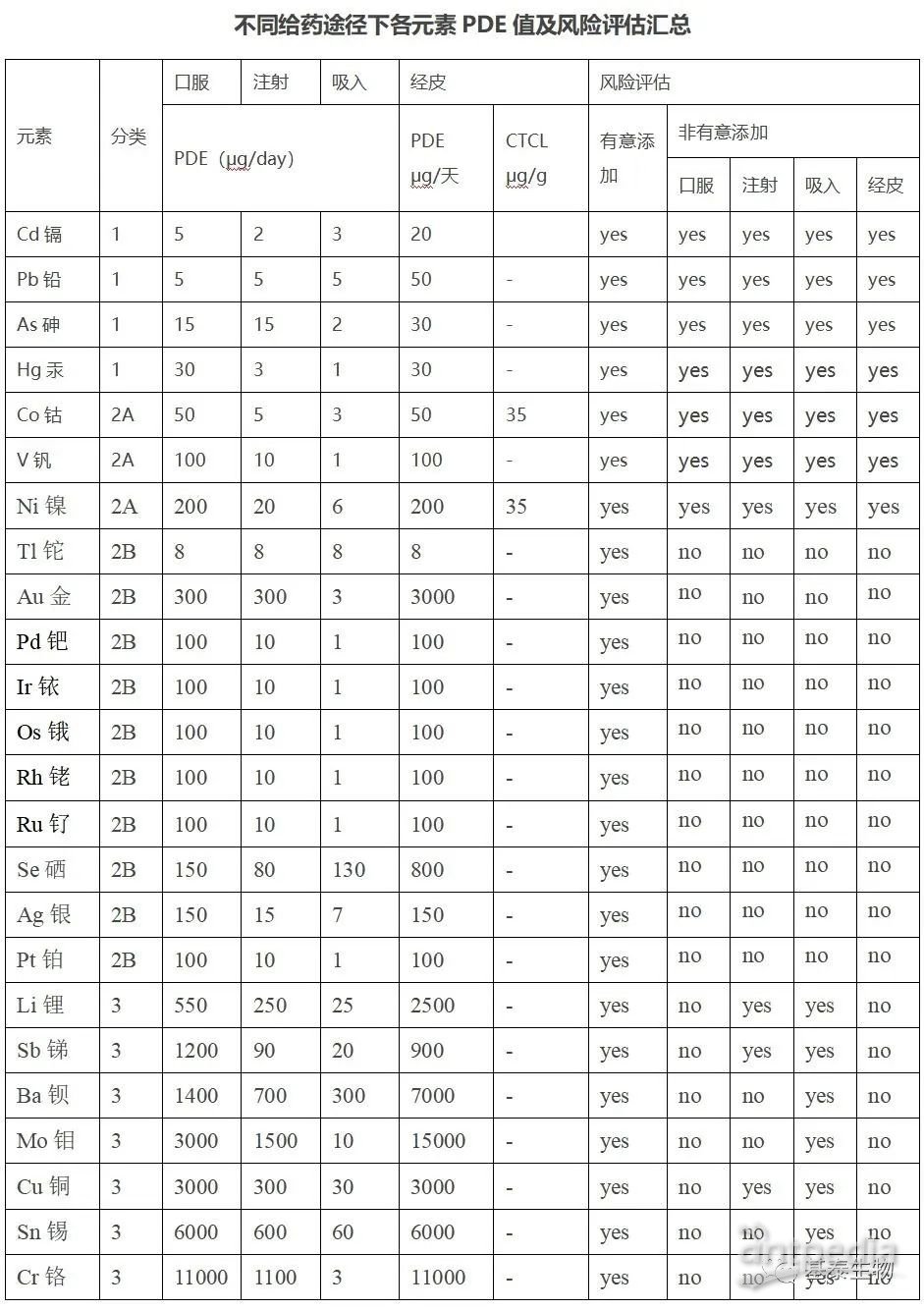
PDE,即每日允许暴露量,以为µg/day为单位。ICHQ3D列出了各个元素杂质的PDE。具体见下表1。同时根据元素的毒性(PDE 值)及其在药品中出现的可能性,ICH指南将元素杂质分为 3 类。但不包括有些元素杂质的 PDE 值还未建的其他元素杂质。
分类 1:元素砷(As)、铬(Cd)、汞(Hg)和 铅(Pb)是人体毒素,在药品生产中禁用或限制使用。这4种元素在所有元素杂质的潜在来源以及给药途径的风险评估中都应进行评估。风险评估的结果将确定那些可能需要额外的控制的组分,在某些情况下额外的控制可能包括对分类 1 中元素进行检测。并不是所有成分都需要检测分类 1 中的元素杂质,仅当风险评估确定需要进行恰当的控制以确保满足 PDE 值时才实施检测。
分类 2:这类元素通常被认为是给药途径依赖型的人体毒素。基于它们出现于药品中的相对可能性,可进一步分成 2A 和 2B 亚类。
a. 分类 2A 元素出现在药品中的相对可能性较高,需包含所有元素杂质的潜在来源以及给药途径(标明的)的风险评估。分类 2A 的元素杂质包括:Co、Ni 和 V。
b. 分类 2B 元素丰度较低以及与其它物料潜在共生的可能性较低,其出现在药品中的概率较低。除非这些元素在原料药、辅料或其它药品成分的生产中有意添加,否则无需进行风险评估。分类 2B 的元素杂质包括:Ag、Au、Ir、Os、Pd、Pt、Rh、Ru、Se 和 Tl。
分类 3:此类元素口服给药途径的毒性相对较低(高 PDE 值,通常>500µg/天),但在吸入和注射给药途径中的风险评估中仍需考虑。除非这些元素是有意添加,否则无需在风险评估中予以考虑。对于注射和吸入给药产品,在风险评估过程中应对含有此类元素杂质的可能性进行评估,除非给药途径的特定 PDE 值高于 500µg/天。此类元素包括:Ba、Cr、Cu、Li、Mo、Sb 和 Sn。
其它元素:由于固有毒性低和/或区域监管的差异性,有些元素杂质的 PDE 值还未建立,应遵从适用于特定元素的其它指导原则和/或地方法规和规范(如:铝导致肾功能损害、锰和锌导致肝功能损伤)或药物制剂的质量考虑(如:存在于治疗性蛋白中的杂质钨)。部分经过考虑的元素包括:Al、B、Ca、Fe、K、Mg、Mn、Na、W 和 Zn。
二、限度控制方法
1、方法 1:药品日摄入量不超过 10 克的药品组分中元素的通用允许浓度限值:
适用于日服用剂量不超过10g的药品。该方法假设药品的日服用剂量为 10g ,且在风险评估中已鉴定的元素杂质(目标元素)存在于药品的所有成分中。采用下面公式计算出各个元素杂质浓度。若在风险评估中,如果药品中所有成分确定的目标元素均未超出方法1的浓度,则所有 这些成分都可以任何比例在该药品中使用。
浓度(μg/g) =PDE(μg / day) ÷日服用剂量(g / day)
例1:以一种口服固体药品为例,该药每日最大摄入量为 2.5g,含有 4 种组分(1 种原料药和 3种辅料,见表1-1)。本药品未超过 10g 的最大日摄入量,故可采用方法1计算的浓度。若每个组分中的金属元素含量均没有超过 PDE 的限度。则药品中每一元素杂质都不会超过 PDE 的限度。
组分日摄入量/g原料药0.2辅料11.5辅料20.5辅料30.3
2、方法 2a:明确药品组分中元素每日摄入量的通用允许浓度限值:
此方法与方法 1 类似,除了日服用量使用实际日最大服用量来计算每个元素的通用允许浓度。基于实际日摄入量,此方法对每个目标元素在药品的每个组分中确定一个以μg/g表示的通用最大浓度的固定值。在风险评估中,如果药品中所有成分确定的目标元素均未超出方法 2a 的浓度,则所有这些成分都可以任何比例在该药品中使用。
例2:仍以例1为例,以一种口服固体药品为例,该药每日最大摄入量为 2.5g,含有 4 种组分(1 种原料药和 3种辅料,见表1-1)。假设经过评估,铅、 砷、
镉、汞需要评估,则按照每日最大摄入量为2.5g分别计算的元素杂质的限度。具体数值见下表2-1。
组分
最大允许浓度(µg/g)铅砷镉汞原料药26212辅料126212辅料226212辅料326212
若各种辅料中的金属元素最大允许浓度均未超过上表限度,则药物中的金属元素浓度不会超过限度。
3、方法 2b:明确药品单一组分中元素每日摄入量的通用允许浓度限值:
分别加和制剂中各组分元素的杂质量。如果风险评估已经确定某个特定的元素不是某个具体组分的潜在杂质,则无需给出该元素在该组分中的定量结果。该方法允许某元素在药品的某些特定组分中的最大允许浓度高于方法 1 或方法 2a 的限度,但需要由在药品其它组分中更低的允许浓度进行补偿。公式可用来表示每种元素在组成药品的每种组分中的特异性限度,以保证满足 PDE 值。
k= 药品中 N 种成份指数;
CK= 成份 k 中元素杂质的允许浓(µg/g)
Mk= 在药品日最大摄入量时成份 k 的质量 2
例3:以一种口服固体药品为例,该药每日最大摄入量为 2.5g,含有 4 种组分(1 种原料药和 3种辅料),各组分日摄入量见下表。各组分中辅料2及辅料3的铅浓度µg/g超过表2-1中铅的最大允许浓度2µg/g。通过下表计算,药物总的PDE为4.01µg/day,小于ICH规定的5µg/day的规定,故药品铅含量符合规定。
组分日摄入量/g铅浓度µg/gPDE µg/day原料药0.21.50.3辅料11.51.21.8辅料20.52.51.25辅料30.32.20.66实际药品中铅PDE µg/day4.01ICH规定铅PDE µg/day6
4、方法 3:成品分析:药物终产品中每种元素浓度是可以测定的。公式 1 可用来以药品最大总日剂量计算某个元素杂质的最大允许浓度。
三、元素杂质水平高于既有 PDE 值可以接受的情况
针对元素杂质水平高于既有 PDE 值(表1)是可以接受的的特殊情况,ICH列了以下几种情况,但明确不限于以下情况:
1、间隔给药;
2、短期给药(比如:30 天或更短);
3、特殊适应症(比如:危及生命的、未满足的医疗需求、罕见病)。
四、元素形态
所谓元素形态,即一种元素的不同物种在特定体系种的分布情况。每种元素杂质都有可能以不同的氧化或配位态存在。例如,砷的毒性表现主要是以无机砷的形式存在,而汞的毒性主要以甲基汞的形式存在,其他形式或价态砷、汞的毒性较弱或无毒。但这些元素的限度是基于毒性最强的形态进行控制的,而元素的测定是通过总量(包括各种形态)来控制的。当测定的元素总量超过限度时,可以测定不同形态的元素杂质。
五、元素杂质控制流程
1、风险评估
通过风险评估来确定需要考虑的元素杂质。需要从有意添加及潜在元素杂质识别两方面进行。同时影响药品种潜在元素杂质水平的因素也需要在风险评估中考虑,这些因素包括但不限于:在后续工艺过程中清除元素杂质的有效性、元素天然丰度(对于非人为引入的元素尤为重要)、对于特定来源的元素杂质浓度范围的先验知识、制剂的组成等。
a. 有意添加元素:在原料药、辅料或其它药品成分的生产过程中有意添加元素(如催化剂)的残留杂质。
b. 潜在元素杂质识别:
l 非有意添加且潜在存在于药品生产所用原料药、水或辅料中的元素杂质。
l 由生产设备潜在引入到原料药和/或制剂中的元素杂质。
l 由容器密封系统潜在浸出至原料药和制剂的元素杂质。
2、分析方法开发及验证
目前USP<233>、<730>介绍了两种元素分析方法,分别是程序1(ICP-AES 或ICP-OES),程序2(ICP-MS),采用专论项下规定参数的程序1及程序2须经药典方法确认,其他参数修改的方法需要进行方法验证。
1)分析干扰
用于微量元素的测定,避免污染是元素首分析要考虑的。潜在的污染源包括不正确清洗的实验室设备和一般设备实验室环境中灰尘等的污染。一个干净的实验室,必须使用专门为微量元素样品处理设计的工作区,用于校准溶液和样品的制备。标准、样品等试剂应尽可能少地暴露在实验室环境中。铬酸不能用于清洗用于ICP-MS分析的实验室器具。此外,ICP-MS检测主要有以下三方面干扰;
光谱干扰:质谱检测中出现的质荷比(m/z)相同的元素,例如由同位素造成的异压干扰,来源于样品本身多原子干扰;
物理干扰:由于样品溶液及校正溶液粘度等方面的差异,导致溶液传输至喷雾器时出现物理干扰,同样由于表面张力等差异,在气溶胶传输至等离子体时,在雾化和电离化过程中出现物理干扰,为了减少上述因素导致的物理干扰,空白、标准及样品溶液的酸组成、浓度必须相同。
记忆干扰:记忆干扰简单的说就是样品检测后,残留在仪器管路部件上的微量元素,从而干扰检测。
2)标准及试剂要求
标准溶液主要用于标化仪器。用于配制样品及标准溶液的所有试剂应不含有元素杂质。可以采用市售的单个元素或多个元素的标准溶液。用于微量分析的标准溶液的有效期是有期限的。一般与浓度、容器的类型、储存条件等都有关系。浓度小于10ppm(W/V)有效期应不得过24小时(来源于USP<730>),除非另有验证数据证明。
检验用水可以用实验室制备的纯化水,浓硝酸、盐酸可以采用市售的专门用于微量元素分析的试剂。
3)标准曲线校正
校正标准溶液:用于校正的标准溶液应每日重新稀释配制。所有校正标准使用50ml聚丙烯离心管,用2% HNO3 / 1%盐酸溶液稀释至50ml。
校正范围:对于每个目标元素,校准范围至少应该是0.5 J到1.5 J。建议对所有元素进行校准小于0.5 J(即0.1 J和/或0.2 J必要的)。
J:元素限值/稀释倍数;
一般情况下,校正曲线的标准溶液浓度应不得少于2个点,对于分析方法经过验证的限度检测,可以用单点校正法。
4)样品溶液的配制
样品溶液的配制方法由样品本身性质决定,主要有以下四种配制方法:
一、直接法:主要适用于液体类样品及其他无需处理样品
二、直接水溶液:适用于溶于水的样品
三、直接有机溶液:适用于溶于有机溶剂的样品。该方法可能需要使用选择性气体。
四、间接溶液法:适用于不溶于水或有机溶剂的样品。推荐总金属提取得到间接溶液,样品采用密闭容器消化等类似方式。
5)样品的消解
一般情况下,大部分样品可以用HNO3和HCl的混合酸消解完全,但是需要注意安全性问题。
6)方法验证
根据USP<1225>分析方法验证要求,方法可分为类别1(含量),类别2(定量杂质及限度测定),类别4(鉴别)。根据USP<1225>对不同类别的要求进行验证。
准确度:
标准溶液:用标准溶液配制含50%J~150J 目标元素的标准溶液;
样品溶液:在样品消化或溶解之前,加标配制含50%J~150J 目标元素的样品溶液;
接受标准:类别1:平均回收率在95.0~105.0%内。类别2:每个杂质的三份不同浓度的元素回收率均在70%~150%之间;
精密度
重复性:样品溶液:6份样品(相同批次),分别按照限度加入标准元素杂质;
接受标准:类别1:RSD%(N=6)应不得过5.0%。类别2:每种元素杂质的RSD%(N=6)应不得过20%。
中间精密度:由不同的实验员,在不同的天或用不同的仪器,按重复性测定;(应至少包括3个不同因素,例如,1个实验员分不同的3天分别按重复性测定;或者1个实验员分2天采用2套仪器,分别按重复性测定;再或者3个实验员用同一台仪器分别测定)
接受标准:类别1:原料药及制剂含量,RSD%应不得过8.0%;类别2:每种元素杂质的RSD%应不得过25%。
专属性
方法应该能够确保在样本种,每个元素都能被明确的检测出。
定量限
定量限QL=10份空白溶液的标准偏差×10。也可用其他合适的方法确定。
接受标准:应至少能够定量准确分析至限度的50%。
线性
配制的线性溶液的浓度应至少包括2点标准溶液及空白(总共3点),线性浓度应包含需要检测样品的浓度,标准曲线应采用合适的统计学方法,例如最小二聚回归。
接受标准:类别1:相关系数R不小于0.995;
类别2:相关系数R不小于0.99;
范围
范围应包括样品的最高、最低浓度具有精密度、准确性、线性的要求。
接受标准:类别1:居中100.0%浓度的验证范围为80.0%~120.0%;非居中浓度的验证范围为:比下限低10%,比上限高10%。含量均匀度:70.0%~130.0%;类别2:验证范围为70.0%~130.0%;
备注:上述验证指标均来自于USP<730>,药典标准为法定最低标准。
耐用性
通过修改实验参数来得到,应符合中间精密度的要求。
3、检测及控制策略的制定
根据风险评估及相关的检测来确定元素杂质水平。可通过3 批代表性生产规模或 6 批中试规模的组分或药品数据,用以建立元素杂质的水平。若该杂质水平小于控制阈(即30%PDE值)可以通过跳检等形式控制,对于大于控制阈但小于限度的元素杂质,可通过日常常规检测的形式进行控制,当然也可以从工艺等各方面进行调整来降低元素杂质水平。针对大于限度的元素杂质,那必须从各个方面进行控制,保证该元素杂质在限度范围内。
07-01 英斯特朗
连载 | 药物一致性评价与粒度分析(三)07-01 欧美克仪器
【仪器百科】LS-909丨干湿二合一激光粒度分析仪07-01 欧美克仪器
标准物质解决方案 | PFASs(全氟及多氟化合物)06-29
第九期阿尔塔有约 | 环境专题【新污染物:PFAS】技术研讨会精彩回顾及提问解答06-29
“绿色技术范式”,分析化学未来发展方向——访中国分析测试协会副理事长、辽宁省分析科学研究院原院长刘成雁教授06-29 转载仪器信息网
华西医院-标准型数显脑立体定位仪、双通道体温维持仪、体式显微镜安装完成06-29 迈越生物
科鉴检测助力2家仪器企业获得首批产品可靠性认证证书06-28 科鉴检测
德国耶拿:锂电池生命周期分析解决方案06-28 德国耶拿
AI已来!生命科学本科教学如何紧跟技术浪潮06-28 Opentrons
盛瀚售后,五星级服务的秘诀是什么?06-28 SHINE
专为汽车制造商打造的柔性解决方案——实现制程控制06-28
西北工业大学-脑立体定位仪安装完成06-28 迈越生物
会议邀请 | 第九届海上检验医师论坛06-28
卓立要闻 | 创新发展ing…6月卓立“大事小情”速览06-28 光电行业都会关注
打造信任合作伙伴!2024年度卓立汉光客户满意度调查开启06-28 光电行业都会关注
如何挑选适用于三阶光学非线性的测量系统?Z扫描测量系统来助力!06-28 光电行业都会关注
招聘启事—中国科学院沈阳自动化研究所微纳光学测量表征技术课题组06-28 光电行业都会关注
谱育科技作为主要完成方 荣获2023年度国家科学技术进步一等奖和二等奖06-28 点击关注→
仪器原理丨顶空仪与吹扫捕集仪科普小知识06-28 天美色谱