生信小工具 | Pathview——通路映射可视化
2022-12-20 01:19:58, 小维 武汉迈特维尔生物科技有限公司
Pathview 是一个用于整合表达谱数据并用于可视化 KEGG 通路的工具,它支持多组学数据映射(基因/蛋白-代谢)。该软件目前有 R包(http://www.bioconductor.org/packages/release/bioc/html/pathview.html)和网页版(https://pathview.uncc.edu/) ,网页版是在R包的基础上,对一些核心功能进行延伸。
Pathview 会先下载 KEGG 官网上的通路图,然后整合输入数据对通路图进行再次渲染,从而对 KEGG 通路图进行一定程度上的个性化处理,并且丰富其信息展示。今天,我们就带大家了解一下Pathview R包的使用方法,让大家能轻松实现KEGG通路可视化。
#1
KEGG PATHWAY 数据库简介
KEGG(https://www.kegg.jp/) 是一个整合了基因组学、生物化学和系统功能信息的数据库,共有17个子数据库,包含不同的类型的信息,有助于研究者把基因及表达信息作为一个整体网络进行研究。

KEGG PATHWAY 是 KEGG 数据库的子数据库,其具有强大的图形功能,包括了代谢、调控、通路、生化、疾病、药物等相关的分子相互作用和关系网络。
▲通路标识符
map,即人工绘制的参考通路,根据已有的知识绘制的、概括的、详尽的具有一般参考意义的代谢图。
ko,高亮标注直系同源基因(KEGG Orthology)的参考通路。ko即KEGG中的基因标识符,ko编号表示一个通路,这个通路是不分物种的,相当于所有物种的这一通路的并集。
ec,高亮标注EC numbers,即酶编号。
rn,高亮标注某个点参与的反应、反应物及反应类型 。
<org>,即物种特异的通路图。
KEGG通路图解读
检索任意一个单基因的代谢通路可以发现其涉及的信号通路是十分复杂的,交叉调控,形成了异常复杂的网络,通路图中存在大量的标注符号,解读通路图的前体是了解这些符号的含义。
KEGG通路图官方帮助文档:
(https://www.kegg.jp/kegg/document/help_pathway.html)
▲通路符号说明

注意:
一个方框有时不单单表示一个基因,有时也表示一类功能基因(或一个基因家族);同样,一个小圆圈有时不单单表示一个代谢物,有时也表示一个代谢物及其同分异构体。
#2
Pathview R包使用介绍
Pathview 是一个用于针对通路进行数据整合和可视化的工具。它将用户数据映射并呈现在相关的通路图上。用户只需要的提供他们的基因或化合物数据,并指定目标通路。该工具可以自动下载KEGG官网通路图数据,解析数据文件,将用户的数据映射到通路,对通路图进行再次渲染,从而对 KEGG 通路图进行一定程度上的个性化处理,并且丰富其信息展示。
Pathview 可以生成两种通路视图(如下),即 KEGG 官网视图(PNG格式)和 Graphviz 视图(PDF格式)。前者将用户数据呈现在原生 KEGG 通路图上,保存了所有关于通路、空间和时间信息、组织/细胞类型、输入、输出和连接的元数据,更自然,更易于阅读。后者使用 Graphviz 引擎对通路图进行布局,更好地控制节点或边缘属性,更好地查看通路拓扑。
2.1 Pathview 的安装
首先,我们要在R里面安装 pathview 包。
一种方法是通过 Bioconductor(https://www.bioconductor.org/install/)安装。Pathview 的安装要求 R 的版本为 v3.5 及以上。
另一种方法是去网上下载 Pathview 的压缩包,https://bioconductor.org/packages/pathview。下载完压缩包之后,进入Rstudio,选择Tools——Install Packages——Browse,找到下载压缩包的位置,安装即可。

2.2 Pathview 的调用
安装好之后,我们需要在 R 里面调用该包,然后进行使用。
查看包的简要概述:
要获得任何函数的帮助(比如pathview),可以使用以下两种形式中的任何一种帮助命令:
2.3 基因数据
输入的基因数据是一个广泛的概念,包括基因、转录本、蛋白质、酶及其表达、修饰和任何可测量的属性。
我们首先加载并查看示例数据,这是一个乳腺癌数据集(表达量数据进行了log2转换)。
2.3.1 单个样本 KEGG 视图
首先,我们观察单个样本在典型信号通路上(“04110”,即“Cell Cycle”)的表达变化。

该例子中的图只有一个图层,在原始图层修改节点颜色,保留原始KEGG节点标签 (节点名)。这样输出的文件大小与原始的KEGG PNG文件一样小,但是计算时间相对较长。如果我们想要一个快速的视图,并且不介意输出文件大小,我们可以通过 same.layer = F使用两个图层。通过这种方式,节点颜色和标签被添加到原始KEGG的额外图层上。原来的KEGG基因标签(或EC编号)被替换为官方基因标签。

2.3.2 单个样本 Graphviz 视图
在上述两个例子中,我们查看了原生 KEGG 通路图上的数据。在这个视图中,我们得到KEGG图上的所有注释和元数据,因此数据更具可读性和可解释性。输出的图形是的光栅图像(PNG格式)。
我们也可以使用 Graphviz 引擎从头布局通路图来查看数据。该图形具有相同的节点和边集,但具有不同的布局。我们对节点和边缘属性有了更多的控制。输出的图形是一个矢量图像(PDF格式)。
Graphviz 视图只需要设置参数 kegg.native = F;同时,当 kegg.native = F时,参数sign.pos生效,该参数用于调整水印的位置("bottomleft","bottomright","topleft" ,"topright")。

该图的主图和图例都在一个图层或者说一个页面中,图例只列出了KEGG边的类型,忽略了节点类型,以节省空间。
如果我们想要完整的图例,我们可以使用两个层来创建Graphviz视图:第1页是主图,第2页是图例。该参数也为 same.layer,设置为same.layer = F。
在Graphviz视图中,我们对图形布局可以有更多的控制,比如可以将节点组拆分为独立的节点,甚至可以将多基因节点扩展为单个基因(注意:在原生KEGG视图中,一个基因节点可能代表多个具有相似或冗余功能的基因/蛋白)。分裂的节点或扩展的基因可能从未分裂的组或未扩展的节点继承边,这样我们就可以得到一个基因/蛋白-基因/蛋白相互作用网络(该功能在网页版不可实现)。
将节点组拆分为独立的节点 split.group = T:

在上面的基础上,将多基因节点扩展为单个基因 expand.node = T:

2.3.3 多个样本
在前面的所有示例中,我们查看了单个样本的数据,这些数据要么是向量,要么是单列矩阵。Pathview 还可以处理多个样本的数据,用于为每个样本生成图形,还可以整合并绘制多个样本的数据到一张通路图中。
一张通路图展示多个样本的信息只需设置参数 multi.state = T:

在这个图中,我们看到基因节点被分割成多个对应于不同样本的片段 (注意颜色块,之前是一个节点一个颜色,现在一个节点是有多个颜色,每个颜色对应一个样本(此处为3个),注意上面代码中的 1:3)。
同样,多个样本也可以生成Graphviz 视图,只需要设置参数 kegg.native = F,此处不再赘述。
2.4 代谢数据
在上面的例子中,我们查看了基因数据在典型的信号通路的可视化情况。除了基因节点外,KEGG通路还有化合物节点。如果我们想研究代谢通路,该工具也可以实现。
在这里,我们先生成模拟的代谢组数据,并加载适当的化合物ID类型进行演示。
2.4.1 单个样本KEGG视图
首先,我们观察单个样本在代谢通路上(“00640”,即“Propanoate metabolism”)的表达变化。代码与基因数据的一致,只是数据集换为代谢数据,数据输入参数由gene.data换为cpd.data。(注意:这两个参数默认值均为NULL,使用pathview函数时,至少有一个需要非NULL。)

Pathview 生成的代谢通路图与原始 KEGG 图相同,只是会将化合物节点放大,为了更好地查看颜色。
2.4.2 单个样本 Graphviz 视图
Graphviz 视图可以更好地显示层次结构。对于代谢路径,Pathview会解析xml文件中的反应条目,并将其转换为基因和化合物节点之间的关系。使用椭圆表示化合物节点。化合物标签是从CHEMBL数据库中检索的标准的化合物名称。由于化学名称是长字符串,我们需要设置参数 cpd.lab.offset 进行自动换行以使它们符合图形上的指定宽度。
当 kegg.native = F 时,cpd.lab.offset 参数才效,其默认值为 ”1” 。

2.4.3 多个样本
同上述基因数据一样,化合物数据也支持处理多个样本的信息,同样也是设置参数 multi.state = T:

同样,代谢物数据的多个样本也可以生成Graphviz 视图,只需要设置参数 kegg.native = F,此处不再赘述。
2.5 基因数据和代谢数据整合
Pathview 为数据集成提供了强大的支持。它可以用来整合、分析和可视化各种各样的生物数据:基因表达、遗传关联、代谢产物、基因组数据、文献和其他可映射到通路的数据类型。
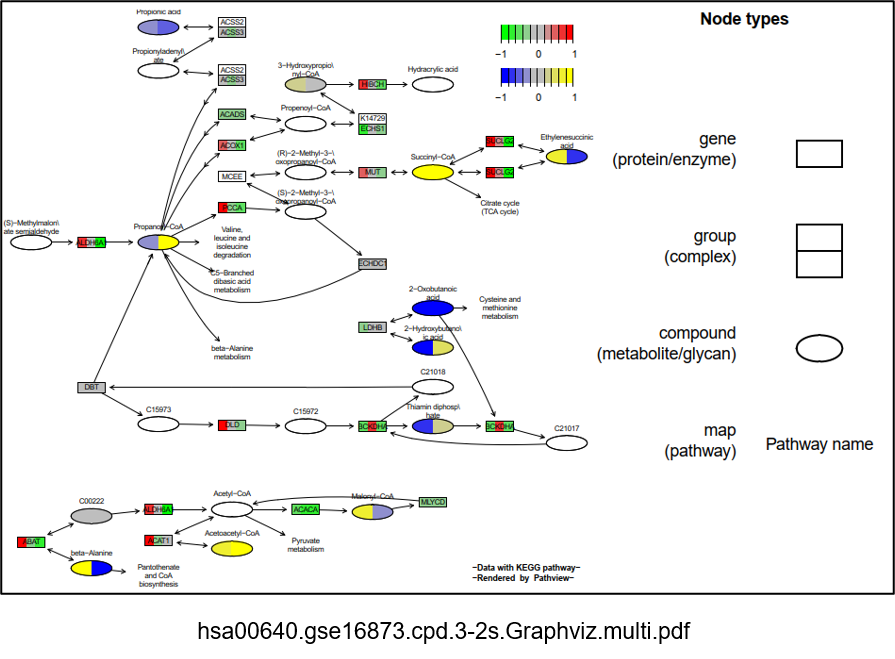
在下面的例子中,基因数据有三个样本 gse16873.d[, 1:3],而代谢数据有两个样本 sim.cpd.data2[, 1:2]。我们可以将所有这些样本包含在一张通路图中。对于这样的多样本数据,可以使用原生 KEGG 视图或 Graphviz 视图。在这些图中,我们看到基因节点和化合物节点被切成多个片段对应不同的样本。由于基因数据和化合物的样本量不同,如果两种数据类型的样本实际上是成对的,即它们的第一列来自同一个实验/样本,我们则可以设置参数match.data 来选择是否进行数据匹配。

下面示例生成 Graphviz 视图:
2.6 离散数据
到目前为止,我们一直在处理连续数据,但实际中我们也经常处理离散数据。例如,根据一些统计数据(P值、Flod Change等)选择显著基因或化合物列表。输入数据可以命名为两个层次的向量,1或0(显著或不显著),也可以是一个更短的显著基因/化合物的名称列表。
在接下来的两个例子中,我们使 gene.data 和 cpd.data 离散或只使 gene.data 离散。
首先生成离散数据:
我们分别查看下sel.genes和sel.cpds的数据结构:
下面的例子使用离散型基因数据和离散型化合物数据:
处理离散型数据需要设置参数discrete,该参数默认值为discrete = list(gene = FALSE, cpd = FALSE)。
参数 limit 用于设置 gene.data 和 cpd.data 数据转换为颜色时的限制值,及颜色标签的范围;默认值为limit = list(gene = 1, cpd = 1)。
参数bins用于设置颜色标签的长度,默认值为 bins = list(gene = 10, cpd = 10) 。

下面的例子使用离散型基因数据和连续型代谢物数据:


99%的代谢组学研究者都在阅读下文(精彩合集,欢迎收藏):
●项目文章 | IF=6.314,TM广靶单组学轻松发文思路挖掘颅内动脉瘤破裂标志物
●激情世界杯,追逐科研梦 | 转录组+代谢组数据挖掘与R语言培训班
●脂肪酸专题:一文了解脂肪酸的分类、合成途径及相关疾病
●蛋白专题合集
●Biomarker专题合集
●空间代谢组合集
●肠道菌群&微生物专题
●生信小工具专题

客服微信:metware888
咨询电话:027-62433042
邮箱:support@metware.cn
网址:www.metware.cn
我就知道你“在看”
06-26 鉴知技术
远离氟污染!开启无氟接触前处理技术新篇章06-26
浑然一体的ChemiSEM技术:集成式扫描电镜成像与 X 射线能谱解决方案06-26
荧光计 VS 分光光度计,倒底怎么选?06-25
最新研究解读:临床基因组测序的普及正助力缩小罕见病诊疗地区间差距06-25
喜讯!奥谱天成·集团及联合创始人刘鸿飞博士荣获新称号06-25
自旋极化子直接观测!无液氦磁体恒温器助力一篇Nature Physics06-25 Dr. Shen
用主成分分析法对市售的含苯基的反相液相色谱柱进行色谱分类和比较06-25 编译:李晓雨
应用方案 | 福立液相采用凝胶色谱法(GPC)测定聚合物分子量06-25
好礼不容错过!第四届药斯卡争霸赛预报名活动,等你来战!06-25 市场部
往期赛事|药斯卡争霸赛往期精选优秀作品(化药篇1)06-25 市场部
往期赛事|药斯卡争霸赛往期精选优秀作品(中药篇1)06-25 市场部
共识速递 | 遗传病SMA最新专家共识发布!06-25
6月19日—21日|CPHI第二十二届世界制药原料中国展—圆满落幕06-25 技尔上海
氨基酸分析仪检测氨基酸纯品中的杂质06-24 大昌华嘉
氨气程序升温脱附测试 (NH₃-TPD) — 脱附能和吸附热的研究06-24 大昌华嘉
润滑油基础油中氮和硫的测试应用---XPLORER NS分析仪06-24 大昌华嘉
大昌华嘉开展Novasina 水分活度仪质量服务月06-24 大昌华嘉
线下培训 | 布鲁克(Bruker)X射线荧光光谱仪用户培训会06-24 大昌华嘉
精彩回顾 | 福立仪器携手纳微科技,共探制药行业新技术06-24