原核转录组知识大放送!一文带你走进原核转录组的世界
2023-07-12 11:36:27, 欧易生物 上海欧易生物医学科技有限公司

同为研究微生物产品的原核转录组,不同于扩增子与宏基因组产品。16S/18S/ITS 统称为扩增子产品,是对特定目标区域进行扩增后再进行测序分析。宏基因组(Metagenome)也称元基因组,是对微生物整个基因片段进行测序的研究方法。要覆盖微生物整个基因片段,所以宏基因组比扩增子需要的数据量更多,有更完整的基因序列因此也可以进行基因功能注释分析。然而,原核转录组是一种单菌落微生物转录本测序,能够获得转录本的结构信息及表达信息,基于NGS平台,通过除核糖体RNA、构建链特异性文库,从基因序列水平和表达水平获得原核生物在某个时期或者在某种环境条件下转录出来的所有转录本(包括mRNA,非编码RNA等)的表达量差异信息及功能特征,找到关键的差异功能基因,揭示微生物不同表型形成的分子调控机制。
原核转录组结构特征:原核生物是多顺反子 mRNA,具有5''、3'' UTR,不具有 poly(A)尾巴,转录与翻译同时进行,寿命期短,具有可变操纵子结构。
1、原核有参转录组
对于有参考基因组的物种,我们优先选用原核有参转录组流程。分析主要包括数据产出统计、参考基因组比对、基因组装与结构预测、基因表达水平分析、差异表达基因分析、非依赖 Rho 因子的终止子预测、sRNA 序列预测、SD 序列预测、GSEA、SNP等内容。
2、原核无参转录组
对于基因组尚未测序和注释的生物体,转录组需要利用测序数据重新组装。基于参考的转录组组装和从头转录组组装都存在许多成熟的计算工具。然而,大多数工具主要是为真核转录组设计的,细菌基因组通常比真核基因组更密集,并且相邻的细菌转录本经常重叠,这使得区分相邻细菌转录本的边界具有难度。多顺反子信息使细菌转录组组装进一步复杂化,特别是当在不同条件下使用操纵子的不同启动子时。此外,真核生物中非编码RNA的模型通常不适用于细菌中常见的小调节RNA。
Rockhopper 2[1] 软件结合两个数据结构,de Bruij 图和 Burrows-Wheeler 指数,使用类似于 RPKM 的测量来估计转录本丰度水平,该测量将转录本的读段数相加,除以转录本的长度和归一化因子。Rockhopper 2 使用更强大的上四分位数转录本表达归一化,使用了高质量的转录组组装,并且在细菌数据组装上优于其他领先的组装软件如:Trinity、SOAPdenovo2。
原核无参转录组分析主要包括数据产出统计、转录本组装、基因表达水平分析、差异表达基因分析、GSEA等内容。除转录本组装外,与原核有参转录组分析结果基本重叠。
*如果没有合适参考基因组,也可以搭配做三代测序的细菌小基因组组装,获得相对更精确、完整的基因组信息和注释用于原核转录组分析。

实验提取样品总 RNA,并进行 RNA 质量检测,去除核糖体 RNA,在 cDNA 二链合成时以 dUTP代替 dTTP,然后连接不同接头,再利用 UNG 酶法将含有 dUTP 的一条链进行消化,只保留连接链不同接头的 cDNA 一链,最后进行 PCR 扩增,使用测序仪进行测序。

原核转录组实验流程

原核转录组分析流程(有参)
1、参考基因组比对
采用 Rockhopper 2 软件进行参考基因组比对分析,该软件采用的比对算法类似 Bowtie2, 基于 BWT(Burrows–Wheeler_transform)数据转化算法构建参考基因组的 FM-index,使比对更加准确快速。
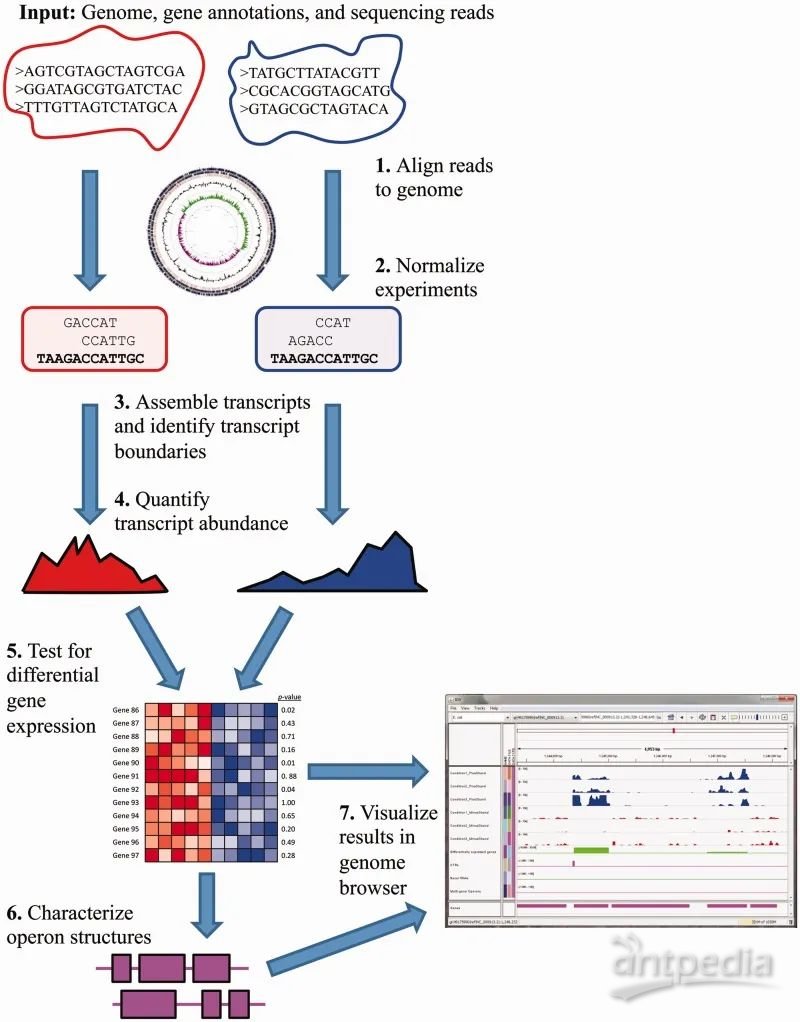
Rockhopper2参考基因组比对分析原理
*对于无参考基因组的细菌微生物,采用 Rockhopper 2软件 denovo 转录组组装,将测序读数与转录本对齐,从而估计转录本丰度水平。

Rockhopper 2 从头转录组序列组装原理
2、基因表达水平分析
用已知的参考基因序列做为数据库,采取序列相似性比对的方法鉴定出各基因在各样本中的表达丰度,使用 Rockhopper 2 软件获取每个样本中比对到基因上的 reads 数以及计算基因的表达量 RPKM 值。RPKM法能消除基因长度和测序量差异对计算基因表达的影响。
PCA 可以从不同维度展现样本间的关系。样本聚类距离或者 PCA 距离越近,说明样本越相似,各组样本分布在二维或三维空间的不同区域,同组的样本在空间分布比较集中。
PCA图
3、差异分析
使用 DEseq2[2] 计算差异基因,其中可视化展示的火山图可以了解差异表达基因的整体分布情况。
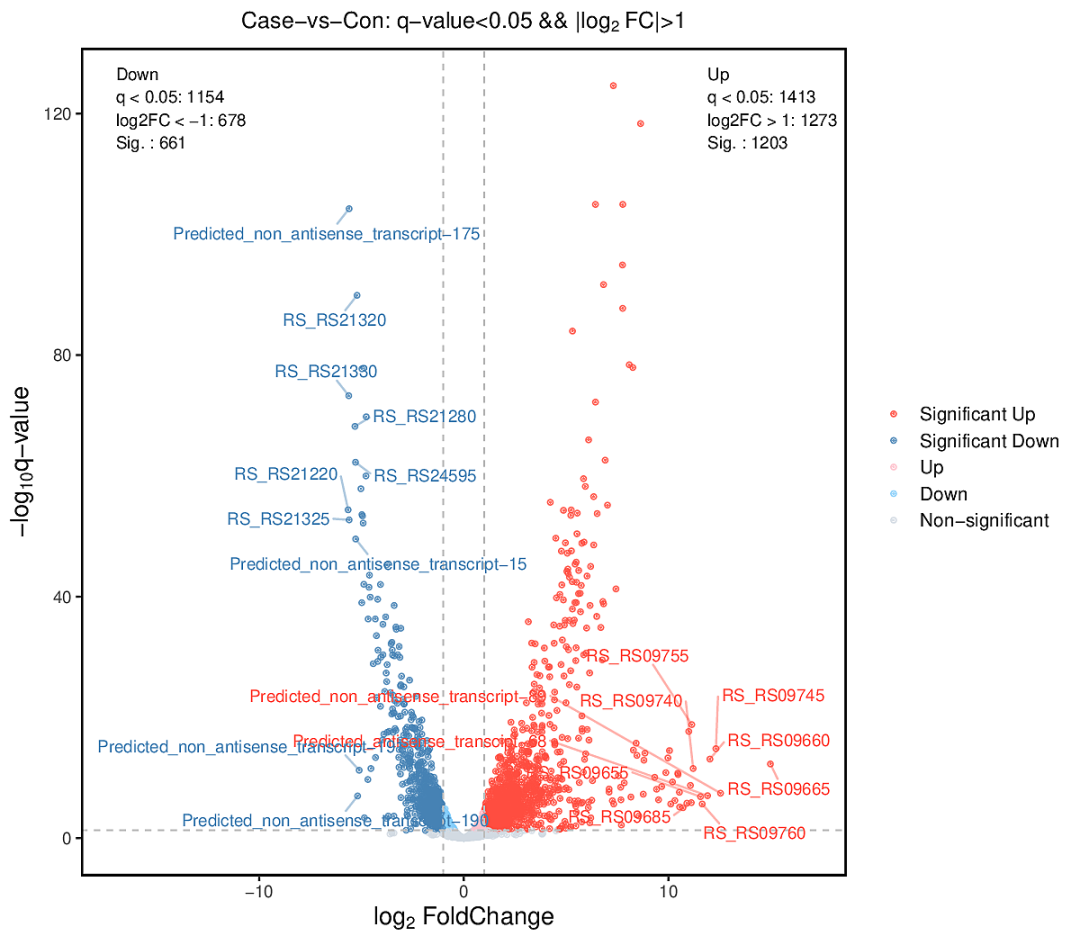
差异基因火山图
4、富集分析
4.1、GO 富集分析
GO 富集分析 top30 (筛选三种分类中对应差异基因数目大于 2 的 GO 条目,按照每个条目对应的 -log10pvalue 由大到小排序的各 10 条)条形图展示如下:

GO富集条形图
4.2、KEGG 富集分析
KEGG是有关 Pathway 的主要公共数据库,利用 KEGG 数据库对差异蛋白编码基因进行 Pathway 分析(结合 KEGG 注释结果),并用超几何分布检验的方法计算每个 Pathway 条目中差异基因富集的显著性。

KEGG富集气泡图
4.3 GSEA
基因集富集分析 (Gene Set Enrichment Analysis, GSEA) [3]是一种用于确定一组预先定义的基因是否在两种生物状态(例如表型)之间显示出统计上显著的或一致的差异的计算方法。其分为三个步骤,分别为计算富集分数、估计富集分数显著性水平和矫正多重假设验证。GSEA 分析是基于全部检出基因进行分析,同时对基因集进行了过滤,默认的标准是基因集最小基因数量为 15、基因集最大基因数量为 500。
GSEA 示例图
基因分组聚类示例图
5、高级分析
5.1、基因组装与结构预测
用 Rockhopper 2 软件将测序结果得到基于序列比对数据获得基因图谱,将该图谱与参考基因注释进行比较,鉴定边界和新基因。
统计文件包括预测非反义转录本数目、预测转录本数目、预测多基因操纵子数目、预测反义 RNAs 数目、差异表达的蛋白编码基因数目、5'' 端 UTR 数目、3'' 端 UTR 数目。
根据鉴定转录起始位点和转录终止位点以及注释文件中的翻译起始位点和翻译终止位点预测得到 UTR 位置信息及其长度信息:
3'' 端UTR长度统计图
5'' 端UTR长度统计图
5.2、操纵子预测
原核生物功能上相关的几个基因往往串联在排列在一起,构成操纵子结构作为一个表达单位,用Rockhopper 2 软件将操纵子预测算法从纯序列特征发展到结合测序实验数据(即计算所得基因表达量),即联合基因间距离和基因表达量相关性两个特征用朴素贝叶斯分类器模型来预测操纵子。对预测得到操纵子进行长度分布、包含的结构基因数目和操纵子链分布进行计算和可视化。
操纵子长度分布图
操纵子结构基因数目统计图
操纵子链分布图
5.3、反义基因预测
新预测的基因中如果基因与已知编码基因重叠或包含,且位于不同的链上,则该基因判定为反义基因,使用 Rockhopper 2 软件预测,反义基因分为三种类型:全部包含(enclosed),3'' 端重叠(convergent)和5'' 端重叠(divergent)。在测序数据来源于链特异文库的条件下,可以预测反义基因位置、类型和数量。
反义基因统计图
5.4、非依赖 Rho 因子的终止子预测
原核生物基因组中有转录终止信号,称为终止子,部分基因转录终止需要辅助蛋白 Rho 因子,但其它基因核心酶本身即可终止转录。不依赖于 Rho 因子的转录终止子具有两个重要结构特征:DNA 顺序有双重对称(dyad),位于 RNA 3'' 端之前 15-20 核苷酸处和 DNA 模板链中有一串约 6 个 A,转录为 RNA 3'' 端的U。双重对称的意义在于其基因能形成发夹结构。采用 TransTermHP[4] 软件预测不依赖于 Rho 因子的终止子序列。
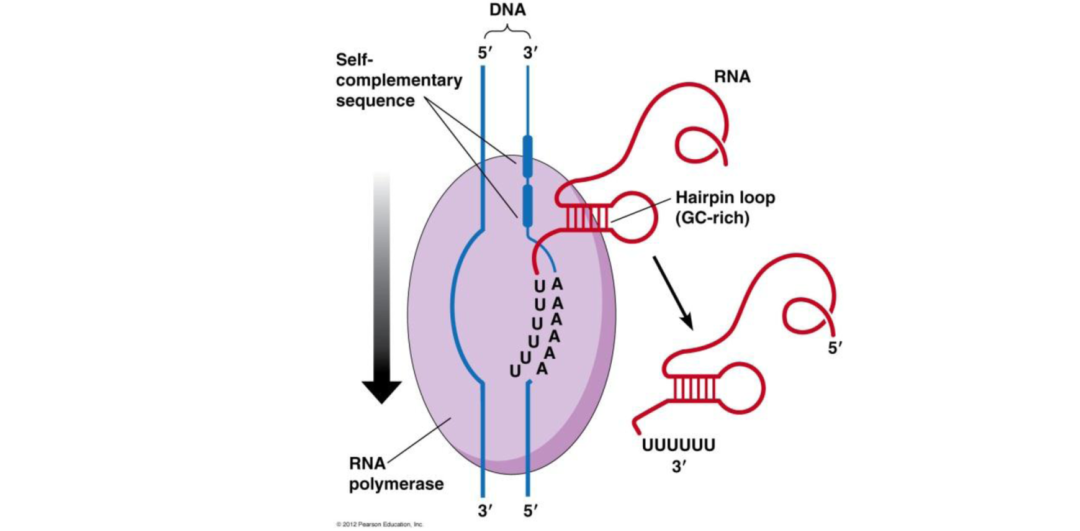
转录终止子示意图
5.5、sRNA 序列预测
原核生物 sRNA 是一类长度在 50-500 bp 的小 RNA 分子,用 Rockhopper 2 软件预测 Novel 基因,RNAFold 分析其茎环结构,进行二级结构预测,使用 IntaRNA[5] 进行靶基因预测,可综合判断 Novel 基因是否为潜在的 sRNA。
5.6、SD 序列预测
SD(Shine-Dalgarno)序列仅存在于原核生物中,SD 序列是一个存在于信使 RNA 上的核糖体结合位点,通常位于起始密码子上游。除引导翻译过程外 SD 序列还有调控翻译效率的作用。采用 RBSfinder[6] 软件预测包含 SD 序列。
SD 序列示意图
5.7、SNP 分析
SNP(Single Nucleotide Polymorphisms,单核苷酸多态性),是指在基因组上单个核苷酸的变异,包括置换、颠换、缺失和插入。以组装好的转录本为模板序列,将原始序列与其进行比对,利用 samtools 软件进行染色体坐标排序、去重等处理,再用 samtools、bedtools 等软件预测样本中的 SNP 和 INDEL 位点。然后利用 snpEff 等软件进行功能注释。为了降低 SNP&INDEL 检测的错误率,使用 QUAL (A quality score associated with the inference of the given allele) 大于等于 20,且 DP(combined depth across samples)大于等于 4 进行过滤结果。对 SNP/INDEl 在基因组上各功能区域的分布进行统计。
01
通过工程改造的冷营养 将半纤维素有效转化为 2, 3-丁二醇:机制和效率
Efficient conversion of hemicellulose into 2, 3-butanediol by engineered: mechanism and efficiency
发表期刊:Bioresource Technology
影响因子:11.889
文章链接:https://www.sciencedirect.com/science/article/abs/pii/S0960852422007829
02
AI-2/LuxS系统在冷藏鲜虾生物保存中的作用:增强植物乳杆菌对营养物质的竞争能力
The role of AI-2/LuxS system in biopreservation of fresh refrigerated shrimp: Enhancement in competitiveness of Lactiplantibacillus plantarum for nutrients
发表期刊:Food Research International
影响因子:7.425
文章链接:https://www.sciencedirect.com/science/article/abs/pii/S0963996922008961
03
蒙脱石对镉诱导大肠杆菌耐药基因的调控机制
Regulatory mechanism of montmorillonite on antibiotic resistance genes in Escherichia coli induced by cadmium
发表期刊:Applied Microbiology and Biotechnology
影响因子:5.56
文章链接:https://link.springer.com/article/10.1007/s00253-022-12075-x
04
比较转录组结合形态生理学分析揭示了嗜热四膜虫捕食诱导嗜水气单胞菌抗噬菌体防御的分子机制
Comparative transcriptome combined with morphophysiological analyses revealed the molecular mechanism underlying Tetrahymena thermophila predation-induced antiphage defense in Aeromonas hydrophila
发表期刊:Virulence
影响因子:5.428
文章链接:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9518995
以上就是原核转录组的相关介绍内容,感兴趣的老师可以联系我们做进一步详细沟通。
参考文献
[1].De novo assembly of bacterial transcriptomes from RNA-seq data. Brian Tjaden. Genome Biology, 16:1, 2015
[2].Love M I , Huber W , Anders S . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2[J]. Genome Biology, 2014.
[3].Aravind Subramanian, Pablo Tamayo, Vamsi K. Mootha. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles[J]. PNAS, 2005
[4].C. Kingsford, K. Ayanbule and S.L. Salzberg. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake[J]. Genome Biology, 2007
[5].Martin Mann, Patrick R. Wright, and Rolf Backofen. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions[J]. Nucleic Acids Research, 2017
[6].Chang TH, Huang HY, Hsu JB, Weng SL, Horng JT, Huang HD. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs[J]. BMC Bioinformatics, 2013
上海欧易生物医学科技有限公司(简称:“欧易生物”),成立于2009年,经过十多年稳健发展,已经成长为拥有“晶准生物”“鹿明生物”“青岛欧易”三家全资子公司,近600名员工的生物科技领域集团型企业。
欧易生物始终秉持着“硬数据 · 好服务”的理念服务于大众。为大生命科学、大健康相关研究领域,以及医药、食品及日化企业的客户,提供从基础研究到药物靶点发现、药理药效及安全性评价、疾病分子标志物筛选、致病菌及耐药菌溯源等相关技术服务,全力加速客户研究与开发进程,提升客户研究与开发价值。

欧易生物携手旗下子公司,实现了中心法则上、中、下游多层组学的串联,从基因组、转录组、表观组、微生物组,到蛋白组、代谢组及近年热门的单细胞&空间多组学技术服务,为科研用户提供全面的创新多组学技术服务。

欧易生物已先后获得上海市科技小巨人企业、闵行区研发机构、闵行区企业技术中心、产权管理体系认证企业等资质。拥有授权发明专利30+项,在受理发明专利50+项,软件著作权150+项。

06-25 技尔上海
氨基酸分析仪检测氨基酸纯品中的杂质06-24 大昌华嘉
氨气程序升温脱附测试 (NH₃-TPD) — 脱附能和吸附热的研究06-24 大昌华嘉
润滑油基础油中氮和硫的测试应用---XPLORER NS分析仪06-24 大昌华嘉
大昌华嘉开展Novasina 水分活度仪质量服务月06-24 大昌华嘉
线下培训 | 布鲁克(Bruker)X射线荧光光谱仪用户培训会06-24 大昌华嘉
精彩回顾 | 福立仪器携手纳微科技,共探制药行业新技术06-24
6月27日 14:30开播 | 电子电器玩具合规性分析的难点及XRF荧光分析应对方案06-24 市场部
2024长春国际光电博览会圆满落幕!06-24 优尼康
展会预告|苏州、深圳双展本周开幕06-24 优尼康
校企携手育英才,合作共赢谋新篇——宁波工程学院与华仪宁创举行共建联合培养基地签约仪式06-24
喜讯|中检维康生物连续五年中标广电计量试剂耗材招标06-24
预告 | LUMEX特邀金牌赞助ICMGP2024第16th全球汞污染物国际会议06-24
应用方案 | 煤中总汞含量测定-DMA3000直接测汞仪06-24 开元弘盛
参会指南|第四届锂离子电池热测试主题研讨会06-24
科诺美 | CPHI China 2024精彩回顾06-21
会议邀请 | 科诺美与您相约“药品质量控制与检验技术大会-济南站"06-21
CFAS2024 | 科诺美诚邀您相聚南京,共话食安!06-21
攻克!AIST 解决碳纳米管的精准测量难题 | 用户访谈06-21 HORIBA
【中药配方颗粒】川芎配方颗粒的检测06-21 DIKMA