新型偶联药物PDC+DAC
2022-10-25 09:54:47, 基泰生物 上海基泰生物科技有限公司


新型偶联药物
PDC+DAC
PDC
多肽偶联药
ADC药物大家已经很熟悉了,那么PDC又是什么呢?
PDC(peptide-drug conjugate)即多肽偶联药物,是一种新型的偶联药物。与ADC药物类似,它也有三个部分构成,分别是归巢肽、连接子和细胞毒性的有效载荷。也是就是说PDC相当于是把ADC药物的运输工具由单克隆抗体换为了多肽。
虽然只是更换了子弹头,但药物的整个原理都得到了更替。在癌症治疗中,有三种靶向方法可以增强癌症治疗的特异性和抗肿瘤活性。其一是抑制肿瘤细胞表达的蛋白质(受体或酶),这就是我们最常见的靶向药,如表皮生长因子受体抑制剂(EGFR-TKI);第二种是使效应分子与肿瘤细胞表面过表达的分子结合,并协同抑制肿瘤细胞分裂,同时提供细胞毒性有效载荷或刺激肿瘤导向免疫反应。双抗类药物、ADC药物和CAR-T都是运用的这种方法;而最后一种就是使用多肽偶联药物(PDC),驱动肿瘤细胞中毒性有效载荷的富集。
PDC的优点
相较于单克隆抗体,多肽更易合成与纯化,这就使得PDC药物在生产、运输和储存上的成本都要更低。
多肽类药物分子量介于小分子药物(<500 Da)和生物制品(>5000 Da)之间,这使它更易穿透肿瘤基质进入到肿瘤细胞当中。而分子量在160KDa的ADC药物会更难进入到肿瘤细胞内部。
由于结构和成分都更加简单,PDC会有更低的免疫原性,即不太会刺激机体发生免疫应激反应。
PDC能够快速被肾脏消除,使其对骨髓和肝脏的毒性更低。
PDC的缺点
稳定性较差,可能会在药物到达作用目标前就被清除。
口服往往会破坏多肽的生物活性,药物往往需要静脉注射给药。而想要将PDC制成可口服的又会增加新的成本。与生物制品相比,肽的循环半衰期更短,往往只有数天至数周,因此需要更频繁给药。
与单抗相比,多肽的组织特异性和肿瘤靶向性都稍有逊色。因此PDC想要实现精准靶向的难度要比ADC更大。这也是导致目前市面上PDC药物较为罕见的主要原因。
PDC药物的结构:
PDC中的靶向多肽种类较多,一般可以分为两大类,分别为细胞靶向肽和细胞穿透肽,常见的细胞穿透肽有Pep-1、Pentratin、PepFact14、Transportan;常见的细胞靶向肽有PEGA、生长激素抑制素类似物、蛙皮素类似物、RGD肽类等。
选择连接体(Linker)是为了允许足够的循环时间,使药物到达其目标细胞。因此,PDC应足够稳定,使肽、连接物和药物在到达靶细胞之前不会被裂解或代谢,并使足够浓度的PDC到达靶细胞,从而提高药物杀灭肿瘤的效果。大多数连接体在系统中就开始被裂解,从血浆(血液)开始,然后是癌细胞的细胞外环境。连接区域常见的官能团大致可分为四类:酶可裂解(酯、酰胺和氨基甲酸酯)、酸可裂解(肼和碳酸盐酯)、可还原二硫醚和不可裂解(硫醚、肟和三唑)。
现研PDC药物中使用的细胞毒性化药,如多柔比星(doxorubicin)、紫杉醇(paclitaxel)、吉西他滨(gemcitabine)和喜树碱(camptothecin),是利用不同机制显示细胞毒性的高度有效的药物。
PDC药物的改进思路
影响药物偶联物疗效的因素有几个,包括靶受体的特异性、细胞摄取、偶联物的半衰期、活性药物或药物代谢物的释放以及药物在靶位点的浓度。而活性药物或药物代谢物从结合物中的释放取决于连接体。为了获得更好的疗效,使用PDC中选择合适的Linker去解决以上问题是一项至关重要的思路。
多肽的化学修饰可以改善其成效性,比如肽订合
多肽侧链的氨基酸改造(如将L-氨基酸更替为D-氨基酸)
制剂优化(如使用渗透促进剂和耐酸涂层)
多肽本身的化学修饰(如将多肽链PEG化或者链接脂肪链)
DAC
降解-抗体偶联物
降解-抗体偶联物(DAC)是一种新型实体,它通过某种类型的化学接头将蛋白水解靶向嵌合体(PROTAC)有效载荷与单克隆抗体相结合(DAC=PROTAC+ADC)
近年来不少研究小组试图通过仿照ADC的设计原理,将PROTAC与单抗(mAb)连接,提高PROTAC体内递送效率。这种叫做抗体偶联降解剂(Degrader-antibody conjugates,DAC)的药物形式可克服未偶联PROTAC分子的以下问题:(1)由于理化性质与DMPK性质(尤其E3连接酶未选用CRBN)导致体内递送效率低;(2)为实现充分的暴露量而采用的复杂的处方;(3)PROTAC非特异性靶向。
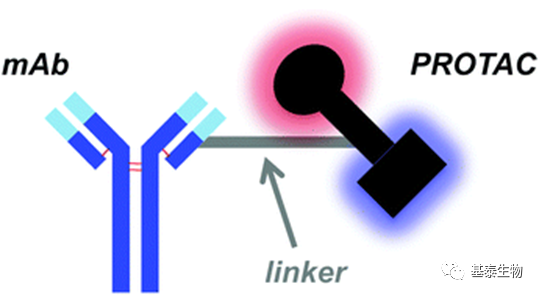
DAC 由 mAb、链接器和 PROTAC 有效负载组成。
将靶抗原的表达与降解有效载荷细胞生物学相匹配。
PROTAC有效载荷必须在溶酶体中稳定,并且能安全逸出。
可能需要 DAR >4,这可能会影响偶联和 DAC 药代动力学。
在开发DAC时,由于PROTAC分子的特殊性,不能简单照搬ADC的递送策略。不同于ADC中使用的广谱细胞毒性小分子药物,DAC中的PROTAC通常只对特定的肿瘤和/或组织或细胞具有靶向活性。因此,抗原的选择不仅需要满足内化DAC并运输到溶酶体中的功能,还需要在PROTAC靶组织(或肿瘤、细胞)上高表达。这种抗原在其他组织或细胞上可能也会有较低水平的表达,只要这类组织或细胞对该PROTAC具有良好的耐受性即可避免脱靶毒性。
由于PROTAC体外活性很多情况下不如小分子药物,因此需要提高DAC中的DAR以发挥药效(即DAC>4)。但是,提高DAR可能导致DAC聚集,对体内药动学(PK)造成不良影响。然而,相较于小分子药物,PROTAC体积更大,亲脂性更强,这种差异会使聚集和PK问题更加严重,需要开发新的连接基团和偶联方法解决上述问题。
许多PROTAC没有可以用于与可裂解连接基团共价结合的位点(氨基)。因此,需要考虑是否需要对PROTAC结构修饰,引入活性位点(但有可能改变PROTAC生理活性);或利用PROTAC现有官能团(如羟基、酚羟基)并开发新的偶联技术。而开发不可裂解基团也需要考虑同样的问题,而溶酶体降解后仍附着在PROTAC分子上的连接基团不得干扰其生物活性。
此外,DAC开发中其他需要解决的问题还包括:
1) PROTAC在溶酶体中的稳定性;
2) PROTAC溶酶体逃逸功能;
3) PROTAC旁观者效应。
后两者受到PROTAC细胞通透性的影响,仍是这一领域当下研究的重点。受细胞通透性影响无法发挥生物学活性的PROTAC可利用DAC中的mAb进入细胞发挥作用,但这类PROTAC旁观者效应预计较小。这种情况下,需要使用不依赖细胞的方法评估该PROTAC生成三元复合物的性能。
目前,DAC仍处于起步阶段,但已经发现了多个具有体外和/或体内生物活性的候选药物。这些DAC结构中的PROTAC分子利用了不同的E3连接酶,靶向不同的目标蛋白。此外,多种新型连接基团以及抗体偶联技术被应用于DAC中。绝大多数DAC分子的DAR(6)比主流ADC(2-4)更大,但这种增加对DAC开发是否为通用规则还需继续研究。多个研究表明,DAC的活性有赖于细胞表面抗原,表明DAC药物能够将对应的PROTAC递送到特定肿瘤和/或细胞中。
基于上述结果,DAC模式的前景光明。但是以下问题仍需要解决:(1)哪些PROTAC分子适合开发成DAC;(2)何种偶联方式能最好得保留(甚至增强)PROTAC分子的生物学活性。
06-27 小 M
有投必奖 | 大家都用 MCE 产品做了啥? (感染领域)06-27 小 M
科研助攻 | 一文讲清:如何破解 PPI 靶点成药难题06-27 小 M
干货分享 | 谈谈天然产物的改构策略06-27 小 M
玉研口鼻暴露系统 | 精准、可控、高通量的动物肺部疾病造模工具,助力创新药物研发06-26 玉研仪器
医药行业指南:电位滴定仪选型攻略来啦~06-26
屹路同行 悦启新程06-26 屹尧科技
徕卡精准空间生物学解决方案 第四弹06-26 童昕、南希
【直播预告】第一届大湾区生物电镜制样讲习班06-26 徕卡显微系统
徕卡常规显微镜历经严苛的ISO9022标准测试06-26 徕卡显微系统
Viventis LS2 Live 光片显微镜发布会06-26 徕卡显微系统
前沿应用 | 经皮无创血糖检测中葡萄糖拉曼峰直接观测06-26 鉴知技术
激光指向稳定在光刻系统应用中的关键作用,及其优化方案!06-26 圈内人都会关注
推陈出新!通微公司携新品亮相本届CPHI06-26 Unimicro
远离氟污染!开启无氟接触前处理技术新篇章06-26
浑然一体的ChemiSEM技术:集成式扫描电镜成像与 X 射线能谱解决方案06-26
荧光计 VS 分光光度计,倒底怎么选?06-25
最新研究解读:临床基因组测序的普及正助力缩小罕见病诊疗地区间差距06-25
喜讯!奥谱天成·集团及联合创始人刘鸿飞博士荣获新称号06-25
欧盟GMP附录1与污染控制策略——更快、更便捷的过程监测创新技术06-25 Sievers分析仪