


Nature | 低亮氨酸饮食改善ER+乳腺癌–他莫昔芬治疗耐药性
美国哈佛医学院贝斯以色列女执事医疗癌症中心Senthil K. Muthuswamy研究团队针对ER+乳腺癌患者接受他莫昔芬治疗产生耐药性的机制进行了探讨。
LLGL1和LLGL2是哺乳动物中调节上皮细胞顶端基底极性建立的支架蛋白,与LLGL1不同,LLGL2在ER+乳腺癌中过表达,并在营养胁迫下促进细胞增殖。LLGL2与SLC7A5和YKT6(膜融合调节剂)形成三聚体复合物,调控细胞表面亮氨酸转运体SLC7A5水平从而促进亮氨酸的摄取和细胞增殖。乳腺癌细胞对激素治疗的耐药性与SLC7A5和LLGL2依赖的营养应激适应有关。SLC7A5对他莫昔芬治疗产生耐药性是必要的,其被认为是乳腺癌激素治疗耐药性的潜在靶点。本研究中发现, LLGL2在ER+乳腺癌中具有促进肿瘤生长而非抑制肿瘤生长的作用,此外,本研究还发现LLGL2对营养胁迫的适应发挥了意想不到的作用。
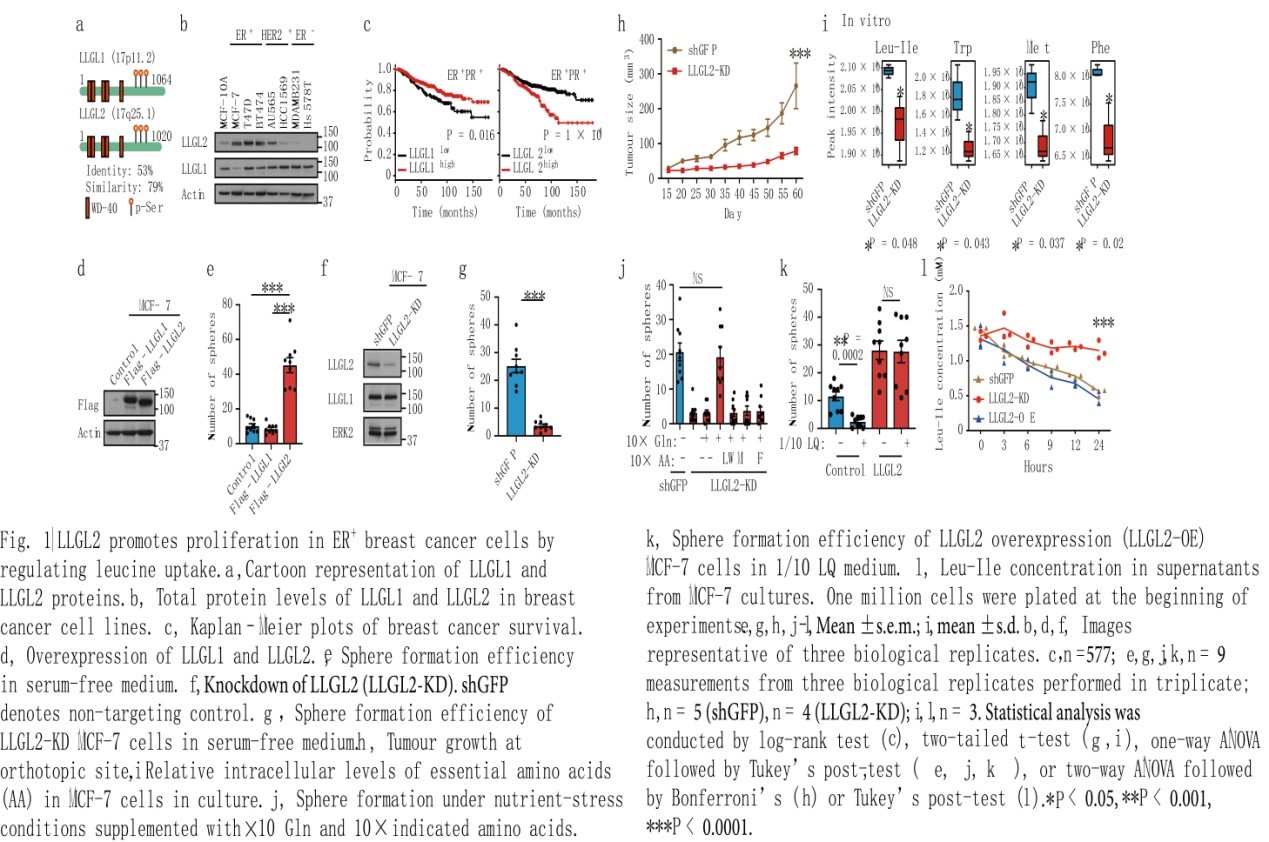
在雌激素受体和孕激素受体(ER+/PR+)表达的细胞中,LLGL2水平是其他细胞的3-5倍(图1a, b)。免疫组化分析显示,ER+乳腺癌组织中LLGL2表达率为70%(24 / 34),而ER-乳腺癌组织中LLGL2表达率仅为14% (5 / 34)。在ER+/PR+乳腺癌患者中,LLGL2 mRNA高表达与较差生存率相关,而在ER-/PR-乳腺癌患者中不相关(图1c),可见ER+乳腺癌患者中LLGL2的表达与不良预后之间存在着意想不到的关联。在正常培养条件下,MCF-7或T47D乳腺癌细胞株中LLGL1或LLGL2过表达并不影响细胞增殖(图1d)。而在无血清条件下,添加表皮生长因子(EGF)和B27的培养基中,过表达LLGL2可促进贴壁和非贴壁细胞的增殖(图1e)。在10%透析胎牛血清(FBS)培养基中(去除氨基酸和其他低分子营养物,保留生长因子),LLGL2-KD细胞增殖受到抑制,表明敲低LLGL2表达导致了细胞增殖对血清中低分子营养物质的依赖,因此添加B27和EGF的无血清培养基可作为营养胁迫条件。
LLGL2如何促进细胞增殖?通过RNA干扰(RNAi)敲低LLGL2(LLGL2-KD)可抑制MCF-7和T47D细胞在贴壁或非贴壁条件下的增殖(图1f, g),而重新表达LLGL2可恢复增殖。此外,敲低LLGL2并不影响细胞的存活,因此无血清培养基中LLGL2是细胞增殖的调节因子。此外,与MCF-7对照组相比,当细胞被垂直注射到免疫缺陷小鼠的乳腺脂肪垫中时,LLGL2在体内的生长被显著抑制(图1h)。因此,LLGL2在培养基和体内都调控ER+细胞的增殖。
为了研究LLGL2如何促进对营养胁迫的适应,通过靶向代谢组学检测LLGL2-KD和对照MCF-7胞内和胞外代谢物的变化,结果显示LLGL2-KD组亮氨酸(leucine)、异亮氨酸(isoleucine)和色氨酸(tryptophan)在培养基和体内肿瘤细胞中均低于对照组(图1i),确定了LLGL2与细胞内必需氨基酸Leu-Ile和Trp水平变化之间的关系。在营养胁迫下,补充过量的Leu能够恢复LLGL2-KD细胞的增殖,说明LLGL2通过促进Leu的摄取来支持细胞增殖(图1j)。过表达LLGL2 (LLGL2- OE)足以促进细胞在LQ(含正常浓度Leu和Gln的十分之一)应激基质中增殖(图1k);超过10倍的Leu和Gln增强了LLGL2-OE细胞的生长,但对照组细胞的生长并没有增强,从而确定了细胞LLGL2水平与LQ调控的细胞增殖间的定量关系。LLGL2-KD消耗胞外亮氨酸的效率低于亲代或LLGL2-OE细胞(图1l),表明细胞LLGL2的水平影响亮氨酸消耗。在10 x LQ培养基中,LLGL2-OE或LLGL2-KD细胞均表现为胞内Leu增加或显著增加的趋势。
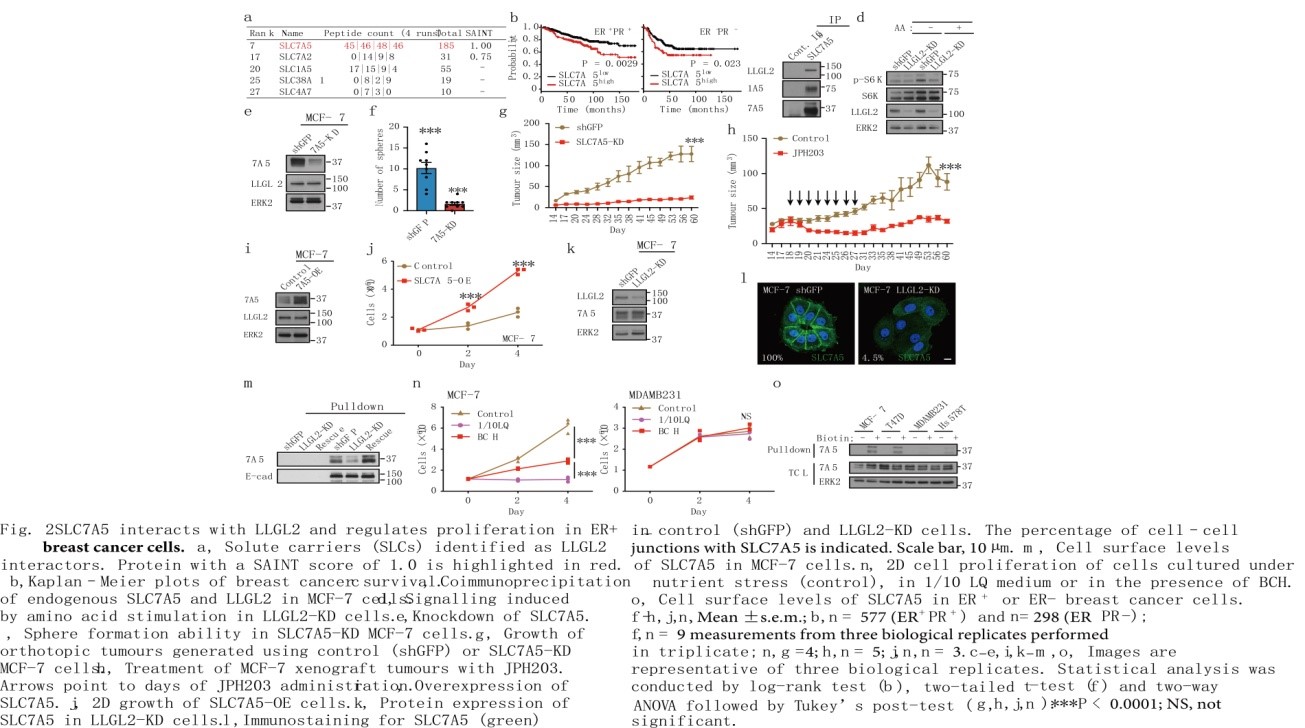
在极化和生理条件下进行近端生物素酰化(BioID)分析鉴定出5种与LLGL2相互作用的溶质载体(SLC)家族蛋白(图2a),结果显示SLC7A5相互作用最强。在ER+/PR+乳腺癌患者中,高水平的SLC7A5 mRNA与较差的临床预后相关(图2b)。SLC7A5也被称为LAT1 (L型氨基酸转运体),是细胞中主要的亮氨酸转运体,在乳腺癌等多种癌症中均过表达。免疫共沉淀分析显示,在营养胁迫条件下LLGL2与SLC7A5和SLC1A5相互作用(图2c)。SLC7A5和LLGL2共定位在细胞连接处,支持了LLGL2与SLC7A5相互作用促进细胞在营养胁迫下增殖的假说。相比对照MCF-7,敲低LLGL2降低了氨基酸刺激mTOR通路靶蛋白S6激酶磷酸化(图2d),提示LLGL2是ER+乳腺癌细胞中氨基酸诱导mTOR活化的调节因子。
在MCF-7或T47D细胞中,敲低SLC7A5(SLC7A5-KD)抑制了细胞在贴壁和非贴壁条件下的增殖,重新表达SLC7A5可恢复细胞增殖(图2e, f)。与对照MCF-7不同的是原位移植的SLC7A5-KD细胞没有形成肿瘤(图2g)。此外,SLC7A5的过表达足以支持细胞在营养胁迫下的增殖(图2i, j),因此SLC7A5是ER+肿瘤细胞在培养液和体内增殖的关键调控因子。
虽然在LLGL2-KD细胞中SLC7A5的总蛋白水平没有受到影响,但SLC7A5在细胞连接中丢失(图2k, l)。根据表面蛋白的生物素标记,LLGL2-KD细胞中SLC7A5的细胞表面水平(而不是E-cadherin)明显低于对照组或恢复的细胞(图2m)。乳腺癌组织免疫组化染色证实SLC7A5在LLGL2high ER+肿瘤中膜定位,而在LLGL2low ER-肿瘤中膜定位不明确。可见在ER+乳腺癌细胞中,SLC7A5定位于细胞连接和细胞膜需要LLGL2。
与LLGL2相比,乳腺癌细胞中SLC7A5的总表达量不随ER状态而变化。使用2-氨基双环-(2,2,1)-庚wan-2-羧酸(BCH)治疗后,ER+细胞的生长明显受损,BCH是一种选择性SLC7A5抑制剂,可破坏亮氨酸转运(图2n)。与ER+相比,ER-乳腺癌细胞表面的SLC7A5水平低8倍(图2o)。而在ER细胞中重新表达LLGL2并没有恢复SLC7A5的表面水平,这表明LLGL2与SLC7A5细胞表面水平的关系是特定于ER+细胞的。因此,与ER+细胞不同,ER-细胞具有在低水平的Leu和低表面水平的SLC7A5中生长的能力。
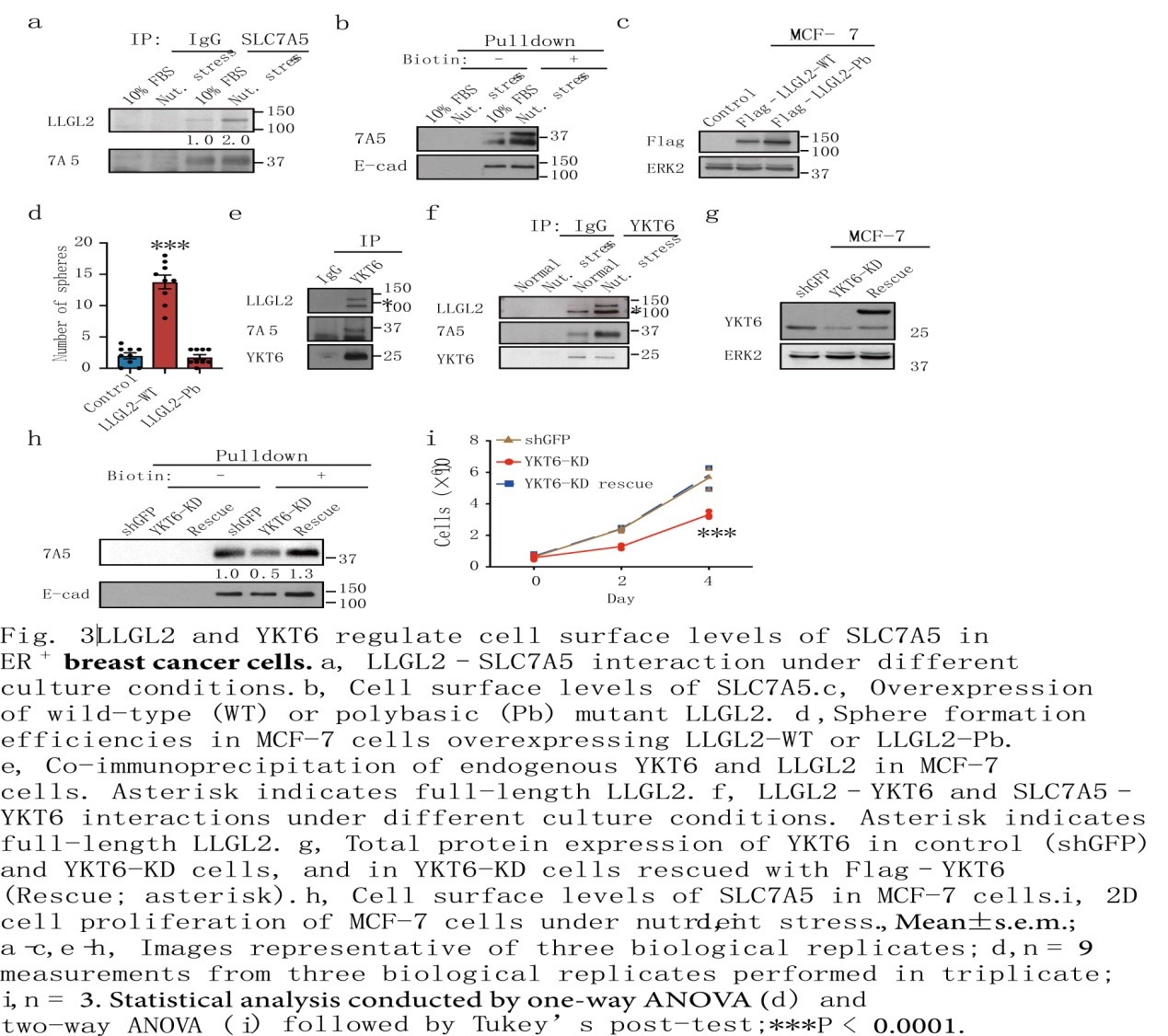
尽管LLGL2和SLC7A5及细胞表面SLC7A5之间的相互作用可以在营养充足的条件下检测到,但它们都是由营养胁迫引起的(图3a, b),表明细胞通过增加表面SLC7A5来适应营养胁迫。LLGL2的N端结构域足以与SLC7A5相互作用。值得注意的是,膜定位缺陷的LLGL2-多元突变体(LLGL-Pb)保留了与SLC7A5相互作用的能力,但未能在营养胁迫下恢复LLGL2-KD细胞的生长(图3c,d)。因此,在细胞质中,LLGL2与SLC7A5相互作用,但这种相互作用不足以促进营养胁迫下细胞的增殖。
为了鉴定LLGL2-SLC7A5复合物的其他成员,作者研究了YKT6(一种可溶性NSF附着蛋白受体(SNARE)蛋白)的作用,内源性YKT6与SLC7A5和LLGL2形成三聚体复合物,以适应营养胁迫(图3e, f)。敲低YKT6可抑制营养胁迫下的细胞增殖,降低SLC7A5的表面水平,抑制LLGL2的表达(图3g i)。因此,YKT6是LLGL2-SLC7A5通路的一部分,其调节SLC7A5表面水平和对营养胁迫的适应。本研究结果发现LLGL2与SLC7A5在细胞质中相互作用并将其转运至膜的机制,其中它与YKT6相互作用以促进膜融合并增加细胞表面的SLC7A5水平。
雌激素(E2)刺激MCF-7和T47D细胞诱导LLGL2的表达增加,这与已知的ER信号靶标细胞周期蛋白D1的增加相似(图4a)。作者研究了LLGL2是否为ER靶点,是否参与E2诱导的细胞增殖。对照组和LLGL2-KD细胞在缺乏雌激素活性的培养基中均不能增殖,相比对照组,LLGL2-KD细胞中E2诱导的增殖能力明显受损(图4b),这说明LLGL2是E2诱导的细胞增殖所必需的。E2刺激足以在LQ胁迫条件下恢复细胞增殖,敲低LLGL2可抑制E2在营养胁迫下恢复细胞增殖的能力(图4c),提示E2诱导的细胞增殖依赖于LLGL2。此外,LLGL2过表达增强了E2诱导的细胞增殖,表明LLGL2水平与E2调控的细胞增殖之间存在定量关系。为了检测LLGL2在营养胁迫条件下调控E2诱导增殖的特异性,作者使用CRISPR Cas9基因编辑技术去除LLGL2基因内含子2中的523-bp ERE(ER结合位点)(图4e),E2刺激未能在LLGL2中缺乏ERE位点的细胞中诱导LLGL2的表达,表明内含子2中的ERE是E2诱导的LLGL2表达的关键介质。ERE的缺失消除了E2诱导的细胞增殖(图4g),表明E2介导的对营养胁迫的适应需要表达LLGL2。
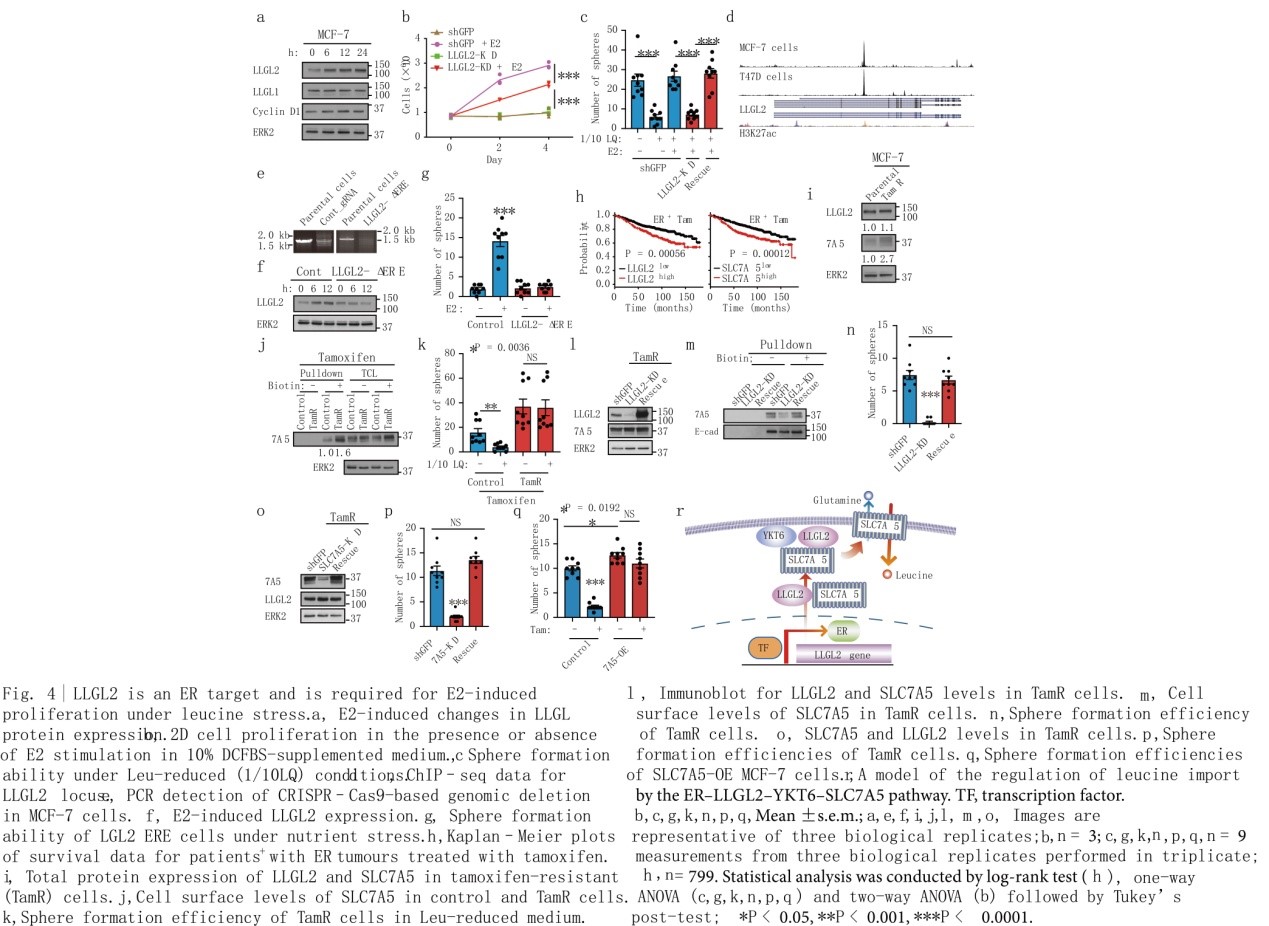
在接受他莫昔芬治疗的799例ER+乳腺癌患者中,LLGL2或SLC7A5的高表达与生存较差密切相关(图4h),表明LLGL2和SLC7A5可能与他莫西芬耐药有关。抗他莫昔芬(TamR)MCF-7细胞中SLC7A5的总蛋白和细胞表面水平显著上调,而LLGL2的水平无明显变化(图4i, j)。不同于对照细胞,TamR细胞对LQ胁迫不敏感(图4k)。LLGL2的下调降低了SLC7A5的细胞表面水平(图4l, m),LLGL2或SLC7A5的下调也足以恢复他莫昔芬对营养胁迫下TamR细胞的敏感性(图4l, n, p),SLC7A5抑制剂BCH抑制营养胁迫下TamR细胞的生长。值得注意的是,SLC7A5在亲代MCF-7细胞中的过表达足以诱导他莫昔芬耐药(图4q)。因此,LLGL2-SLC7A5是一种以前未被报道的他莫昔芬耐药调控者(图4r)。
小结
本研究发现细胞极性蛋白LLGL2和亮氨酸转运之间存在着意想不到的关联,结果表明LLGL2通过帮助癌细胞战胜营养胁迫,促进了肿瘤生长。E2对营养胁迫的适应和LLGL2氨基酸转运途径的调控促进了他莫西芬的耐药,是针对乳腺癌内分泌治疗耐药性的潜在靶点。
参考文献
Yasuhiro Saito, Lewyn Li, ,Myles Brown& Senthil K. Muthuswamy,et al. LLGL2 rescues nutrient stress by promoting leucine uptake in ER+ breast cancer[J]. Nature(2019). https://doi.org/10.1038/s41586-019-1126-2.
微信公众号:麦特绘谱
Tel:400-867-2686
Email : marketing@metaboprofile.com
Web: www.metaboprofile.com

| 标签: | 低亮氨酸 ER+乳腺癌 他莫昔芬治疗 麦特绘谱 |