
南京集思慧远生物科技有限公司
诚信认证:
工商注册信息已核实! 扫一扫即可访问手机版展台
扫一扫即可访问手机版展台
砷暴露对小鼠肠道宏基因组和代谢谱的响应
背景:人类肠道是一个极其复杂、多样和庞大的微生物群落-肠道微生物群。肠道微生物群在代谢过程、能量产生、免疫和认知发育、上皮稳态等方面发挥着重要作用。然而,肠道微生物群的组成和多样性很容易受到外界因素的影响,这就增加了接触有毒环境化学物质导致肠道微生物群改变或失调的可能性。砷的暴露影响全世界的大量人口,并且与许多疾病有关,包括癌症,糖尿病和心血管疾病。
目的:研究了砷暴露对肠道微生物组成及其代谢状况的影响。
方法:采用16S rRNA基因测序和质谱代谢组学分析相结合的方法,研究砷暴露对肠道微生物群的功能影响。
结果:16S rRNA基因测序结果表明,10 ppm砷暴露4周后,砷对C57BL/6小鼠肠道微生物组分有明显的干扰作用。此外,代谢组学图谱显示了一种并行效应,许多肠道菌群相关的代谢物在多个生物基质中被干扰。
结论:砷的暴露不仅改变肠道微生物群落的丰度水平,而且在功能水平上也严重干扰其代谢状况。这些发现可能为肠道微生物群的微扰及其作为接触砷导致或加重人类疾病的潜在新机制的作用提供新的见解。
材料和方法
动物和处理:
无特定病原体的C57BL/6雌性小鼠20只(6周龄,体重20±3g)。平均分为两组。
两组小鼠在同样的环境条件下饲养,供给相同的食物和水。
饲养到8周龄后,处理组:饮用水中注入无机砷(亚砷酸钠,10ppm)持续四周;对照组:依旧供给清水。
在整个实验过程中,每天评估小鼠腹泻,脱水和身体状况恶化的情况。
用砷4周后,用二氧化碳对小鼠实施安乐死并进行尸体剖检。对肝脏(左侧,内侧,右侧和尾状叶)和结肠(远端,横向和前期结肠)的多发区域的炎症,水肿,上皮缺损,高血浆和发育异常等指标进行评估。病理评分未显示对照组和砷治疗组小鼠之间存在任何显着差异,也未观察到体重,死亡率和食物摄入量的任何显着变化。
16SrRNA测序数据分析:
Illumina Miseq;PE150bp。
使用QIIME软件(http://qiime.org)对原始配对的fastq文件进行质量过滤,解复和分析。
UCLUST软件(http://www.drive5.com/uclust)用于选择OTU。
使用核糖体数据库项目(RDP)分类器(http://rdp.cme.msu.edu)选择来自每个OTU的代表性序列组用于每个OTU的分类学鉴定。
GreengenesOTU(4feb2011构建)参考序列(97%序列相似性)用作RDP分类器的训练序列。
代谢组学的样品处理:在小鼠安乐死前一天,尿液收集容器周围放置干冰的代谢笼收集尿液样本,以防止在收集期间氧化或降解代谢物。另外从个体动物收集粪便颗粒。尸体剖检期间收集血浆样本。
代谢组学分析:LC-QTOF-MS
代谢组学数据的数据处理:MassHunter软件进行数据格式的转换。
对具有显著变化的分子特征的确切质量进行了人类代谢组数据库(HMDB;http://www.hmdb.ca/),METLIN(http://metlin.scripps.edu)和KEGG数据库的搜索。
结果
探索肠道微生物组功能变化的工作流程:
本实验采用16 SrRNA测序和代谢物分析相结合的方法,研究砷暴露对肠道微生物群及其代谢谱的影响。检查肠道微生物组变化与代谢组移位之间的相关性,以确定砷暴露对肠道微生物组的功能影响。
砷诱导的肠道微生物组变化:
图1A显示了在科水平上鉴定到的肠道细菌,每种颜色代表单个细菌家族。图1B中列出了肠道细菌组分的分类和倍数变化(p<0.05),使用多元统计分析可以很容易地区分由砷引起的肠道微生物组模式的差异。如图1C中的PCoA图所示,对照和处理的动物分离良好。与PCoA图一致,通过带有算术平均值(UPGMA)的未加权对组方法的折叠β多样性和层次聚类分析表明,所有对照和处理的动物都聚集在它们自己的组中,如图1D所示。
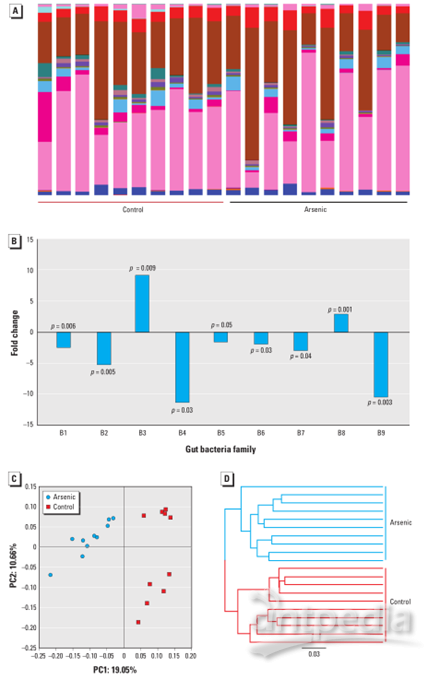
图1:(A)通过16SrRNA测序显示对照和砷处理小鼠的科水平的肠道微生物组成谱(每种颜色代表一个细菌科)。(B)与对照相比,砷处理小鼠中显着扰动的肠道细菌的倍数变化和分类分配(C)通过主坐标分析区分对照和砷处理小鼠的肠道微生物组模式。(D)UPGMA的分层聚类分析表明对照和砷处理的小鼠聚集在它们自己的组中,UPGMA距离树构建在0.03的距离。
砷引起肠道微生物代谢谱的改变:
大量肠道细菌及其代谢产物在粪便中的结合,为评估肠道微生物群的功能变化提供了理想的生物样本。图2A显示,砷的暴露扰乱了肠道微生物群的代谢特征,分别有146条分子特征的增加和224条的减少。如图2B所示,使用代谢组图谱可以很容易地区分对照组和砷处理组,并对照组和砷处理动物进行了很好的分离,无交叉重叠。图2C中的分层聚类热图也显示了每个组中检测到的分子特征的相似聚类模式。
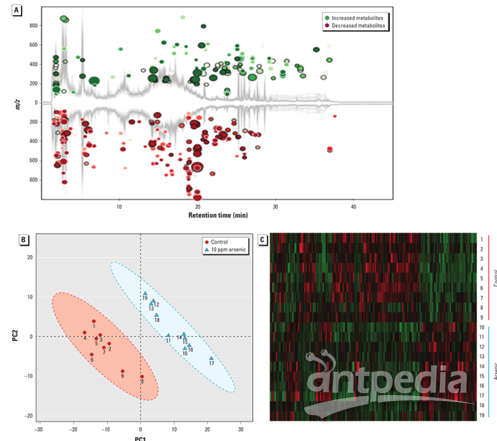
图2:(A)砷暴露使小鼠粪样代谢改变,其中370种分子特征与对照组相比有显著变化。(B)通过PCA将对照物与代谢物分布的砷处理小鼠分开。(C)使用具有1.5倍变化的分子特征构建的分层聚类热图(p<0.05)在各个组内显示一致的聚类模式
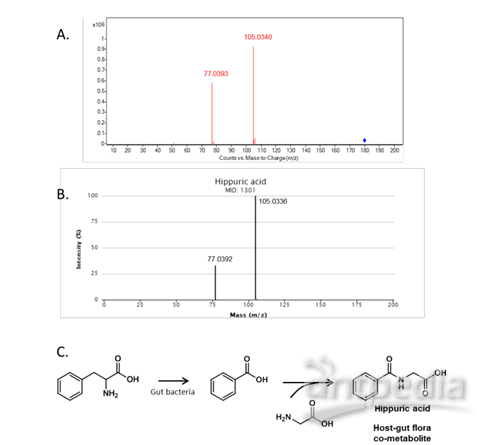
图S4:在血浆样品中鉴定出显着改变的代谢物(与对照相比倍数变化>1.5;p<0.05)。在15分钟时m/z=180.0658的MS/MS谱将该代谢物鉴定为马尿酸(A:尿代谢物;B:数据库中马尿酸的MS/MS谱);通过苯丙氨酸代谢合成嘌呤酸,并且已经将其鉴定为宿主-肠道菌群共代谢物(C)。(进一步的代谢途径分析表明,马尿酸是宿主-肠道菌群共代谢物,其丰度与不平衡的肠道微生物组成成分密切相关。)
肠道微生物组与代谢物的相关性
为了探索肠道微生物组变化与代谢物扰动之间的功能相关性,通过计算Pearson相关系数生成相关矩阵(图3A)。肠道微生物群的变化与代谢产物的变化有明显的相关性(p>0.5或<-0.5,p<0.05)。图3B列出了几种典型的肠道菌群相关代谢物,它们与特定的肠道细菌高度相关,以显示肠道微生物群和代谢物群之间的功能相关性。
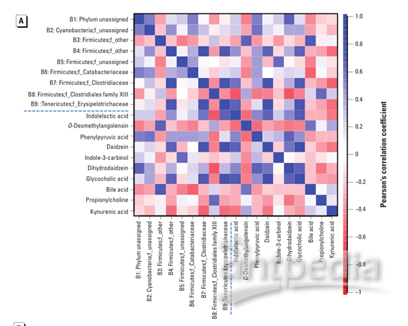
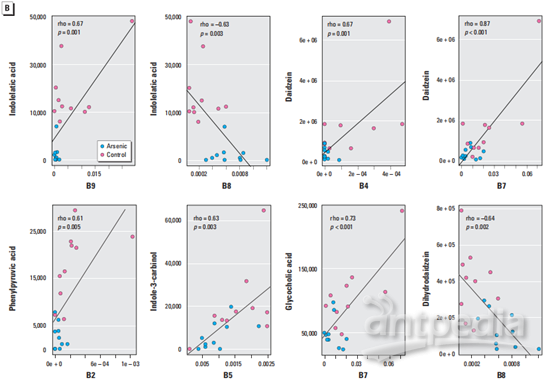
图3:(A)相关图显示了扰动的肠道细菌家族和改变的粪便代谢物之间的功能相关性(差异代谢物与微生物群落相关性分析)。(B)散点图说明了改变的肠道细菌家族的相对丰度与一些典型的肠道微生物群落相关代谢物(包括含吲哚的化合物,异黄酮代谢物和胆汁酸)的质谱强度之间的统计关联(特定代谢物与特定微生物种属相关性分析)。
结论
文章结合16S rRNA测序和代谢组学来分析砷对小鼠肠道微生物组及其代谢组成的影响。测序结果显示,砷暴露显著改变了肠道微生物区系组成,而代谢组学分析表明,接触砷后,许多参与不同代谢途径的代谢产物受到严重干扰。此外,相关性分析发现,一些肠道细菌家族与肠道微生物相关代谢物的改变高度相关。
总之,这些数据表明,砷的暴露不仅使肠道微生物群在丰度水平上受到干扰,而且还改变了肠道微生物群的代谢特征,从而支持假设:肠道微生物的扰动可以作为一种新的机制,通过该机制,砷暴露会导致或加剧人类疾病。此外,这些调节的肠道菌群相关代谢物可能是潜在的生物标志物,有助于探索砷和其他多种环境化学物质对肠道微生物群落的功能影响。
参考文献:Lu K , Abo R P , Schlieper K A , et al. Arsenic Exposure Perturbs the Gut Microbiome and Its Metabolic Profile in Mice: An Integrated Metagenomics and Metabolomics Analysis[J]. Environmental Health Perspectives, 2014.
原文链接:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3948040/


