
诚信认证:
工商注册信息已核实! 扫一扫即可访问手机版展台
扫一扫即可访问手机版展台
科研快讯 | MGISEQ-2000助力广东省新冠病毒基因组流行病学的首次揭示
4月16日,广东省公共卫生研究院、广东省疾病预防控制中心及牛津大学共同在Cell 在线发表题为“Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China ”的研究论文。研究第一作者广东省公共卫生研究院陆靖、刘哲以及牛津大学和爱丁堡大学的Louis du Plessis和Verity Hill,通讯作者为牛津大学的Oliver G Pybus和广东省疾病预防控制中心的柯昌文。该研究结合了宏基因组测序(MGISEQ-2000,PE100)和多重PCR扩增子测序方法,从广东省受感染的病例个体中获得53个新型冠状病毒基因组序列,首次揭示了中国广东省新型冠状病毒基因组流行病学。
研究背景 广东省新冠病毒地方性流行现状
研究对中国人口最多的广东省(1.13亿人口)进行了分子监测。截至2020年3月19日,广东省通过不同的监测策略共开展了160万余次新冠病毒实验室检测,确诊了COVID-19病例1388例。在这1388例新冠病毒的阳性样本中,1014例可能有湖北接触史,334例与本地传播有关。在与本地传播有关的334例病例中,53%的病例(181例)与家庭成员之间传播有关,60%的病例来自深圳和广州。
研究方法 新冠病毒基因组序列测定
研究从65例来自广东省新冠病毒实验室检测结果为阳性的病例中获得了79份临床样本(如图1所示),基于多平台(MGI/ILMN/ONT)进行了宏基因组测序和多重PCR扩增子测序,获取53个新型冠状病毒基因组序列。研究发现(如图2所示),相同Ct值下基于华大智造MGISEQ-2000的宏基因组测序方法完成的新冠病毒基因组数据更容易获得较为完整的基因组,而多重PCR扩增子引物测序产出的序列在拼接过程中存在少数覆盖范围较差的区域。
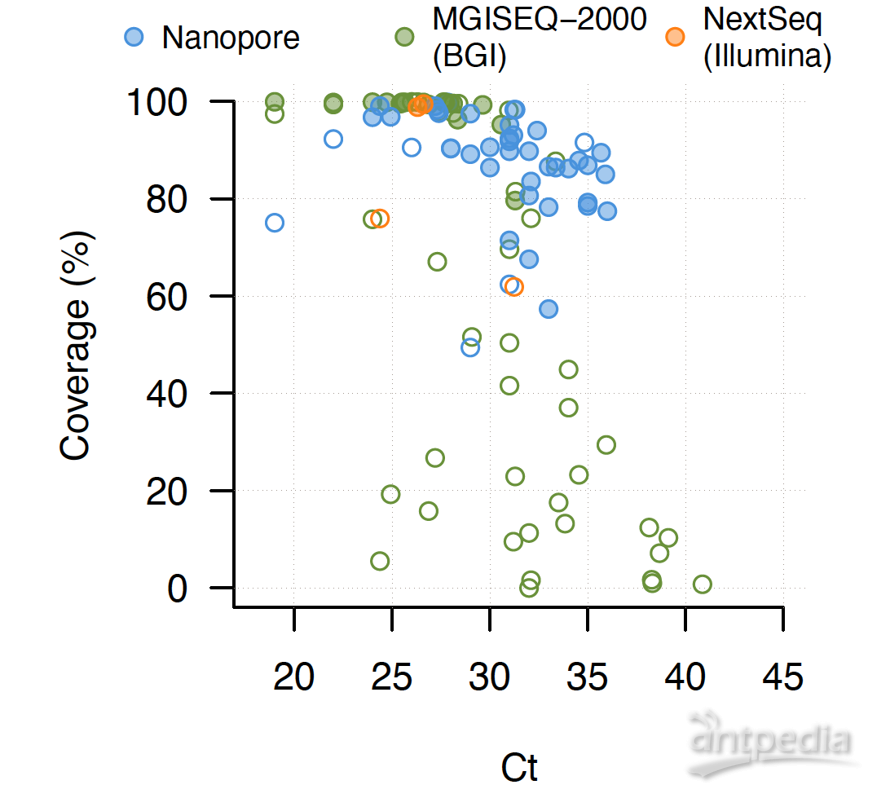
图1 79份临床样本核酸检测Ct值分布图
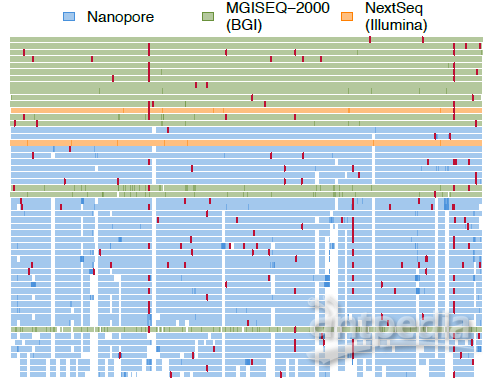
图2 79份临床样本的104次测序运行表现(按新冠病毒基因组覆盖度的完整程度降序排列)
红点表示每条序列中的单个核苷酸的差异位置
结果和结论 本地有效管制措施很重要
无论是基于遗传距离还是分子钟的进化树都可以看到多数广东的病例序列散发在整个进化树中,中间穿插着其他地方或国家的序列。这个结果表明:多数广东的COVID-19病例为不同区域的输入性感染,而不是本地传播感染导致。
在疫情流行初期的新冠病毒遗传变异低,多数系统发育聚类并不稳定,但仍可以发现5个本地序列形成的簇(A-E)具有较高的后验值支持(>80%),仅从进化关系上可能认为这些序列对应的病例是本地传播形成。但通过流行病学调查可以发现cluster中的多数序列都有湖北旅居暴露史。这一结果表明,如果缺乏疫情区的序列数据,需要非常小心的解释从进化关系上看到的结果。这些观察到的广东聚集序列可能是由多个同源或高度同源的输入病例序列形成。
研究首次结合基因序列数据及流行病学数据揭示了广东省内新冠病毒基因组流行病学特征。最重要的是,研究揭示了像在珠三角这样人口密度高并且有频繁外来疫情输入的地区,仍然可以通过大范围的分子监测,积极的病例追踪加严格的隔离措施来阻断本地的传播链条。随着当前广东省从其他国家输入性病例数量的增加,仍需要持续进行并加强新冠病毒疫情的监控。
原文链接:https://marlin-prod.literatumonline.com/pb-assets/products/coronavirus/CELL_CELL-D-20-00921.pdf
参考文献:Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China.DOI: 10.1016/j.cell.2020.04.023
