
诚信认证:
工商注册信息已核实! 扫一扫即可访问手机版展台
扫一扫即可访问手机版展台
DIA与AI的结合—NM又发新文(IF:28.467),通过神经网络和干扰校正实现高通量蛋白质组的深度覆盖
前言
2019年11月,英国剑桥大学生物化学系和米尔纳治疗学研究所(Department of Biochemistry and The Milner Therapeutics Institute, University of Cambridge)等多家机构在一区期刊nature methods(IF=28.467)发表题为“DIA-NN:通过神经网络和干扰校正实现高通量蛋白质组的深度覆盖”的文章,该文章作者提出了一种方便的集成软件包DIA-NN,它利用深层神经网络和新的量化及信号校正策略来处理DIA蛋白质组学的实验结果。DIA-NN提高了传统DIA蛋白质组定性和定量的能力,特别在高通量应用方面具有快捷的优势,与快速色谱方法结合使用时能够对蛋白质实现准确的深度覆盖。
基本信息
英文标题:DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput
中文标题:DIA-NN:通过神经网络和干扰校正实现高通量蛋白质组的深度覆盖
软件版本:DIA-NN(1.6.0)、OpenSWATH18、Spectronaut、Specter、Skyline
平台:QExactiveTM HF(Thermo Fisher Scientific ™ )、TripleTOF ® 6600 (SCIEX)
材料:酵母蛋白提取物、人脐带血血浆、酶解的人K562细胞裂解物、Hela细胞蛋白提取物、大肠杆菌蛋白提取物
影响因子:28.467
发表期刊:nature methods
运用技术:DIA蛋白质组学

|
研究背景
蛋白质组学提供了细胞中基因组和代谢组之间的功能联系,并且迅速在个性化医疗和新兴的数据驱动生物学领域中占据重要地位。然而,蛋白质组固有的复杂性阻碍了数据的产生。在基于质谱的(bottom-up)蛋白质组学中,这种复杂性导致肽检测的随机性,降低了蛋白质组样本的检测深度。这些问题的一个通用解决方案是通过蛋白质组或肽段预分组份来降低样品的复杂度,从而提高了蛋白质组覆盖度,但代价是时间和资源的增加,以及样品之间的变异性增加。
替代性的DIA蛋白质组学技术方法,可以减少随机因素。在DIA分析体系中,质谱仪不是以数据依赖的方式(DDA)选择丰度最高的母离子(即具有特定质/荷比的肽)进行进一步分析,而是对一组预设的前分离窗口进行循环扫描;并且,在目的质量范围内,所有的母离子都会被碎裂进入二级质谱检测。DIA工作流程显示出很高的重复性,并且最近被证明在Orbitrap型质谱仪的单针分析中比传统的DDA模式在蛋白质检测深度上表现更好。
然而,由于DIA技术产生的数据非常复杂性,DIA数据集的计算处理仍存在问题。一个关键的问题源于这样一个事实:在DIA中,每个母离子并不是只产生一个峰,而是每个母离子会由于碰撞诱导解离产生大量碎片离子并形成数据信息量极其庞大的二级质谱图。此外,由于未碎裂的母离子的干扰,这些质谱图通常是高度复杂的。在短色谱梯度与复杂样品两种情况同时出现的情况下,干扰会进一步放大,限制了DIA-MS在高通量蛋白质组学中的应用。鉴于增加样本通量可减少批次效应,加速研究进程,从而能够对大量样本进行研究,因此必须克服这些挑战。
为了应对这些挑战,作者开发了DIA-NN,一个处理高度复杂DIA数据的软件包。
研究思路
|
实验结果
1.什么是DIA-NN
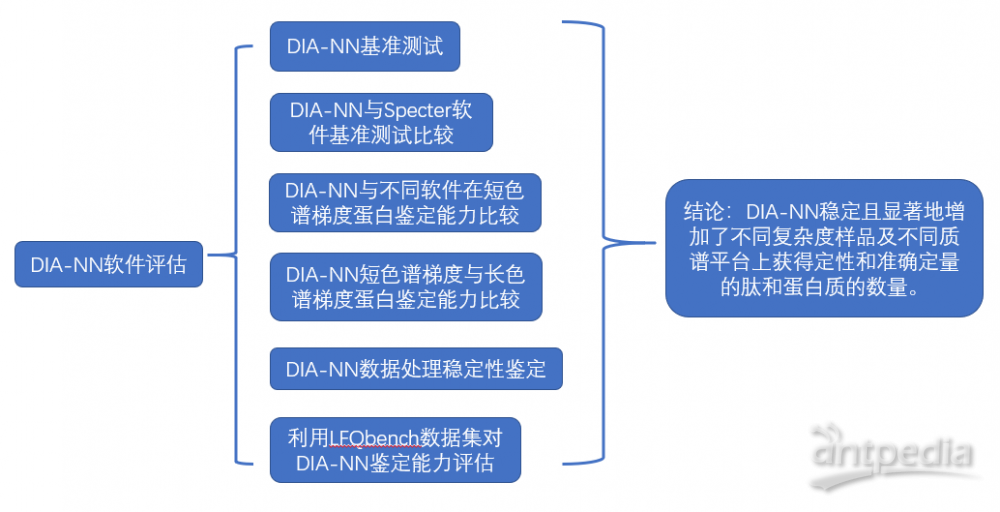
DIA-NN使用深度神经网络(DNNs)来区分真实信号和噪声,并使用新的量化和干扰校正方法。DIA-NN流程是完全自动化的(图1),拥有直观的图形界面和命令执行工具,结果以简单的文本格式展示。DIA-NN使用内源肽(如iRT)进行保留时间校准。DIA-NN还可自动执行质量校正,并自动确定搜索参数(如保留时间窗口和质量提取精度)。这消除了需要为每个特定数据集做优化的繁琐过程。
|
图1 | DIA-NN工作流程示意图
提取每个母离子及其所有碎片离子的谱图(谱图为示意图,不同片段对应不同颜色)。然后对假定的洗脱峰进行评分,并选择“最佳”峰(用星形标记)。然后检测并去除潜在的干扰肽。匹配的母离子峰匹配通过使用DNNs集合计算q值并且从碎片离子洗脱曲线中去除干扰及定量,最后定性及定量蛋白质。
2.DIA-NN的工作流程
DIA-NN的工作流程首先是基于一组母离子的以肽为中心的方法(对每个母离子的多个碎片离子进行注释),这些母离子可由谱库提供或由DIA-NN在电脑中从蛋白质序列数据库(无库模式)自动生成。然后DIA-NN生成一个阴性对照库(即诱饵母离子),为每个目标或诱饵母离子提取谱图,并识别由母离子和碎片离子洗脱曲线组成的假定洗脱峰,该洗脱峰位于假定的母离子保留时间附近。每个洗脱峰由一组反映峰特征的分数来描述,评分项目包括碎片离子的共洗脱、质量准确性或检测到的离子与参考(库)谱图之间的相似性。总之,DIA-NN在工作流程的各个步骤中计算了73个峰值分数(图2,部分展示)。然后,使用线性分类器的迭代训练为每个母离子选择最佳候选峰,该线性分类器允许计算每个峰的单个分数。

|
图2 | 用DIA-NN对假定洗脱峰进行评分(部分展示)
评分项目包括:离子共洗脱(MS2水平)、离子共洗脱(MS1水平)、同位素离子共洗脱、总信号、测量的碎片相对强度、质量精度(MS2)、保留时间(RT)、洗脱曲线形状、其他假定洗脱峰的存在、母离子库的特征值。
3.DIA-NN如何提高定性准确性
尽管这种以肽为中心的搜索灵敏性很高,但仅靠此可能导致错误的定性和不可靠的量化,因为数据中的一个假定洗脱峰可以用作多个具有相近m/z值的一个或多个碎片离子的母离子的检测证据。该问题的一个解决方案是利用以谱峰为中心方法的优点,为每个谱峰选择单个最佳匹配母离子。DIA-NN评估最初保留时间相同的多个母离子之间的干扰程度,如果认为干扰足够显著,则仅报告那些打分最高的母离子数据,通过严格的错误发现率(FDR)阈值提高定性能力。
|
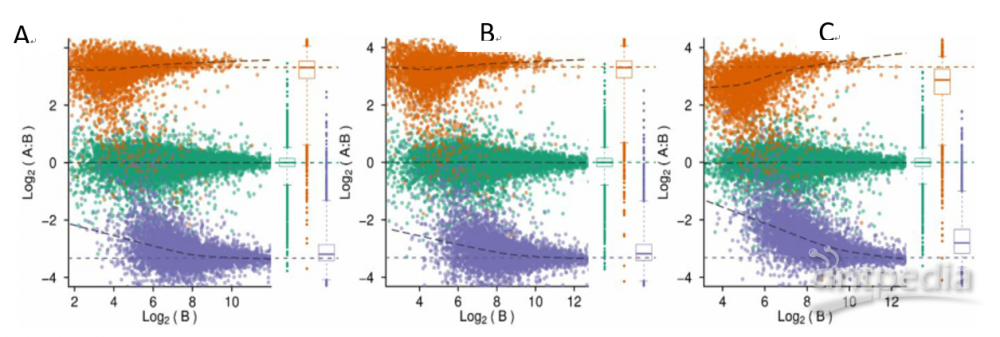
图3| DIA-NN定量算法的性能
A:默认参数下DIA-NN的LFQbench肽数量比率图;B:禁止量化片段的交叉运行选择;C:禁止从碎片的洗脱曲线中去除干扰。n=15743(人),3755(酵母),4997(大肠杆菌)
|
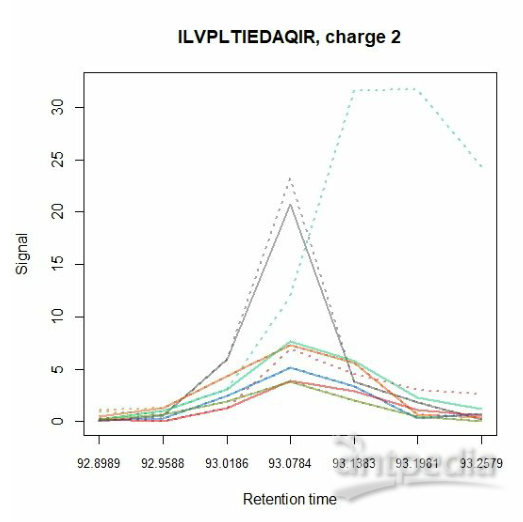
图4 | 用DIA-NN去除碎片洗脱曲线中的干扰
在干扰校正之前(虚线)和之后(实线)绘制大肠杆菌肽的谱图(LFQbench试验)。DIA-NN假定第三个库片段(m/z 487.299,蓝色洗脱曲线)代表肽的真实洗脱曲线,并使用其提取的洗脱曲线来去除其他片段提取的洗脱曲线的干扰。
5.DIA软件的基准测试
作者使用专门为测试DIA软件而创建的公共数据集对DIA-NN进行了基准测试。首先,使用具有不同色谱梯度长度(从0.5 h到4 h)的纳米流动液相色谱耦合QExactive HF质谱仪(Thermo Fisher Scientific)对胰蛋白酶酶解的HeLa全蛋白质组体系进行鉴定能力评估。用OpenSWATH、Skyline和Spectronaut Pulsar(Biognosys)对相同的数据进行处理,并对其进行了调整,以获得最佳结果。蛋白质组基准中的一个关键问题是,每个软件以不同的方式计算FDR,因此不能直接比较所获得的肽段数。事实上,最近已经证明,即使对诱饵母离子生成算法进行简单的更改,也会造成分析工具报告的内部FDR估计值减半或加倍。因此,我们使用无偏双物种谱库方法评估有效FDR,其中(非冗余)人类样本中分配的诱饵母离子被计算为假阳性。与其他测试工具相比,DIA-NN的识别性能更好,在严格的FDR阈值下观察到最大的差异(图5)。
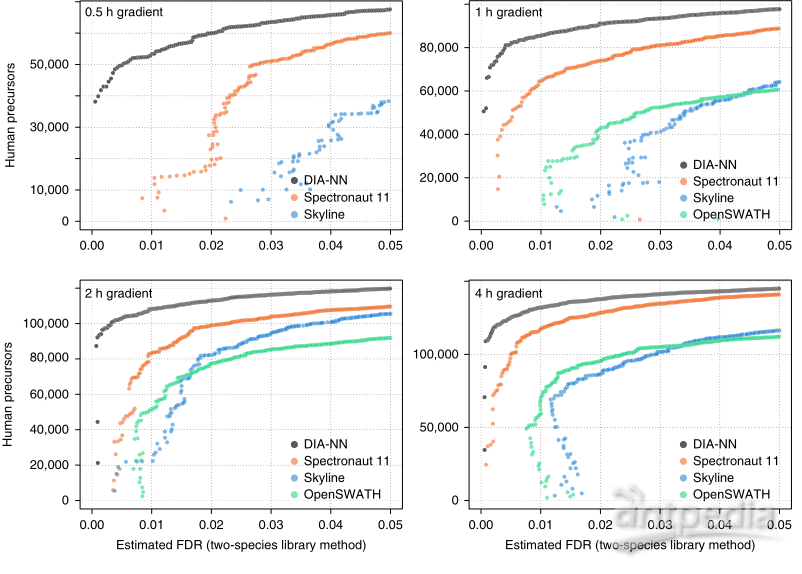
|
图5 | DIA-NN对胰蛋白酶消化的Hela细胞的鉴定能力(QExactive HF,0.5-4 h梯度长度)
图上的每个点对应于诱饵母离子,X轴值反映在各自的得分阈值处的预估FDR,Y轴值是在该阈值处识别的目标(人)母离子的数目。出于技术原因,OpenSWATH没有对0.5 h的采集组份进行分析。
在微流液相色谱-三重耦合6600质谱仪(SCIEX)上用19分钟色谱梯度快速分析K562人细胞株全细胞胰蛋白酶消化液时,也观察到类似的改进。即使使用基于Orbitrap的谱库,DIA-NN也能识别超过35000个肽段(图6),这比几年前Orbitrap质谱上2小时色谱梯度所获得的肽段数量还要多。

|
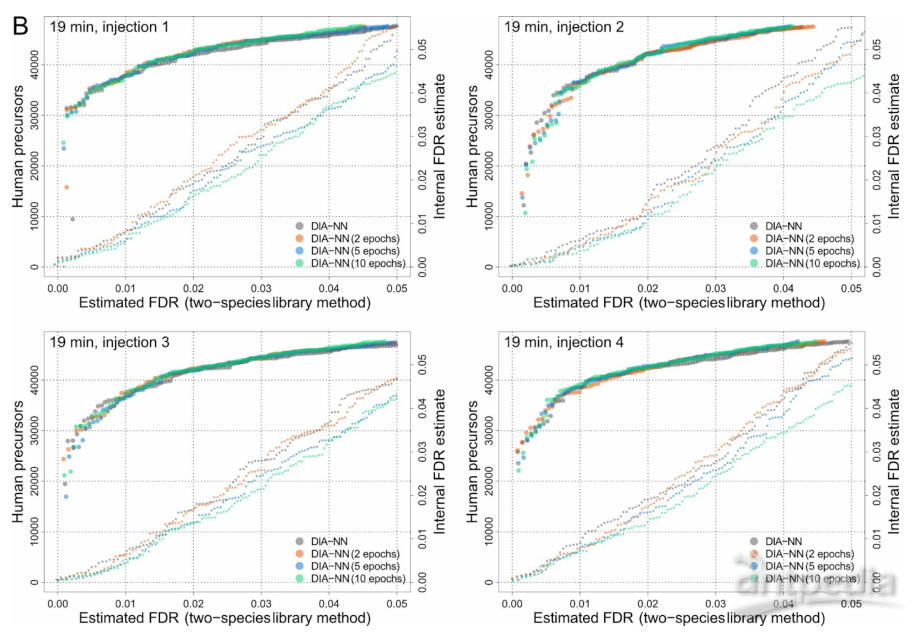
|
图6 | 高通量蛋白质组学中非项目特异性谱库进行有效的肽段鉴定
A:利用高通量蛋白质组学中的非项目特异性谱库进行有效的肽鉴定。DIA-NN在严格的FDR(<2%)下明显优于Spectronaut,识别出了数倍于Spectronaut的肽段,并证明了更精确的内部FDR预估。B:单一训练时期(默认参数)不太可能导致过度拟合。
6.DIA-NN与Skyline或OpenSWATH在短色谱梯度的鉴定能力的比较
重要的是,DIA-NN能够对短色谱梯度进行可靠的蛋白鉴定和蛋白质组深度覆盖。在所有考虑的FDR阈值下,用色谱分离相同的样品,DIA-NN从0.5小时色谱分离中识别出比Skyline或OpenSWATH从1小时色谱分离中鉴定到的肽段数量更多(图5),并且在1%FDR条件下的2小时采集和0.5%FDR条件下的4小时采集中获得数据最多。
7.DIA-NN与Spectronaut在短色谱梯度与长色谱梯度下对蛋白鉴定能力的比较
为了验证DIA-NN在短色谱梯度下的鉴定效果,作者将其分析结果与长色谱梯度下的结果进行了比较。DIA-NN在0.5小时鉴定的前50000个肽段中,49694个在1小时、2小时或4小时(在1%FDR下,由Spectronaut计算)被Spectronaut确认。此外,DIA-NN在0.5h时鉴定的数量与Spectronaut在低干扰下4h采集中鉴定的数量更为相似,而不是Spectronaut在0.5h时鉴定的数量,这可能是由于Spectronaut在0.5h时鉴定错误的次数更多(图7A、B)。这些错误鉴定可能是由于在密度图X轴2.5处出现的混合峰有关(图7C)。
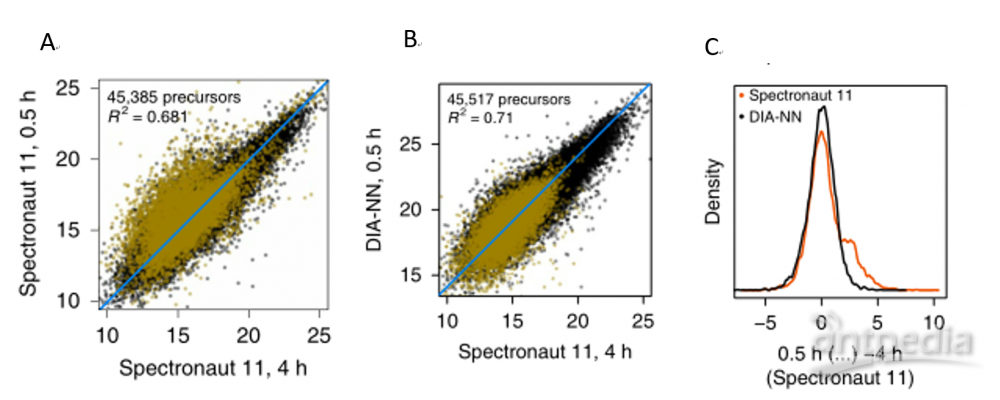
|
图7 | DIA-NN在短色谱梯度与长色谱梯度下的鉴定效果
0.5 h采集(Spectronaut(A)或DIA-NN(B)鉴定到前50000个肽段)和4 h采集(Spectronaut鉴定到前100000肽段)鉴定的log2-肽段数量图。在0.5 h仅由Spectronaut(总共8379)或DIA-NN(总共8511)识别的肽段以黄色突出显示。C图绘制了用Spectronaut或DIA-NN鉴定的0.5 h 时的log2-肽段数量与Spectronaut鉴定的4 h时的 log2-肽段数量差异(中心)的分布密度图。
8.DIA-NN与Specter软件的基准测试比较
作者将DIA-NN与Specter软件进行了基准测试,但由于软件固定的FDR阈值,因此我们使用了不同的基准测试。DIA-NN也显示出比Specter软件更好的鉴定能力(图8)。
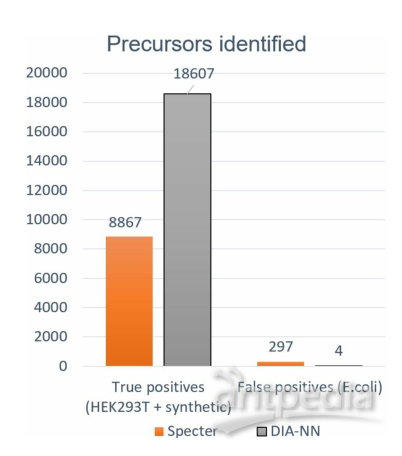
|
图8 | DIA-NN与Specter的鉴定能力比较
9.DIA-NN对高通量数据处理的稳定性鉴定
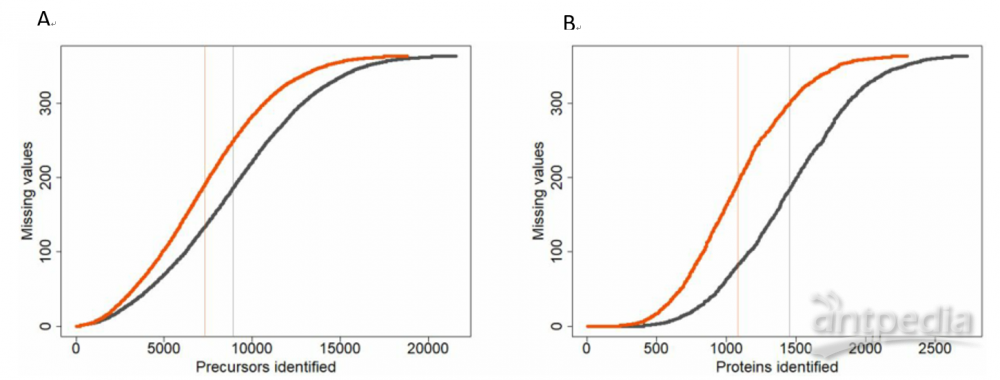
作者使用364个酵母样本(包括非必需激酶的缺失)证明了DIA-NN对高通量数据处理的稳定性佳(图9)。分析这些样本,DIA-NN在传统PC上花费大约3小时处理数据集(图10)
|
图9 | 深层神经网络的使用不会对鉴定的稳定性产生负面影响
364个酵母样本中的缺失数量根据肽段(A)和蛋白质(B:仅考虑唯一识别的蛋白质)的数量绘制,黑色曲线表示深层神经网络分类器启用,橙色表示禁用。定性数量平均值用垂直线表示。
|
图10 | DIA-NN的处理速度
10.利用LFQbench数据集对DIA-NN鉴定能力进行评估
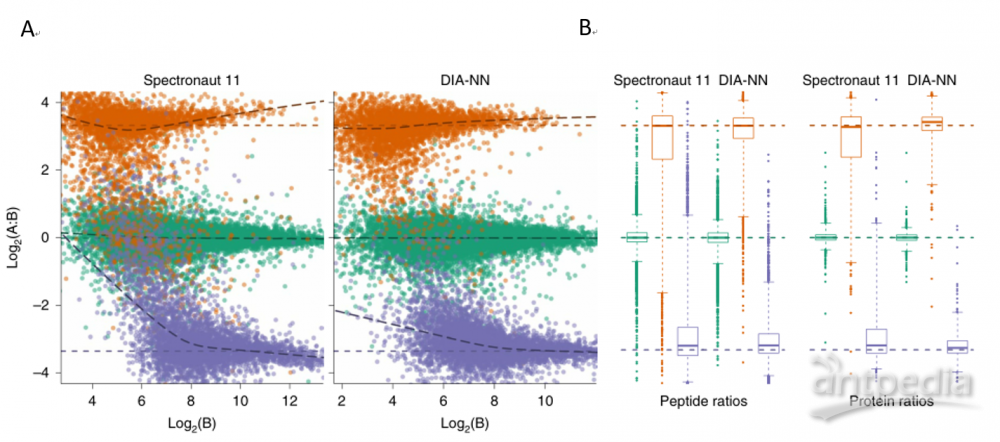
作者使用专门设计用于比较DIA软件的LFQbench数据集,根据三种裂解物(人、酵母和大肠杆菌)之间的已知肽段比率去评估定量准确性。与Spectronaut相比,DIA-NN对酵母和大肠杆菌肽和蛋白的定量精度更高(图11)。此外,DIA-NN提高了人类肽和蛋白质的CV中值(分别为5.6%和3.0%),而Spectronaut为7.0%和3.8%。此外,我们以无库模式在LFQbench数据集上对DIA-NN与Spectronaut进行了基准测试,发现DIA-NN比Spectronaut定量出更多的人类肽和蛋白质,并且具有更高的精度。
|
图11 | 用LFQbench检测DIA-NN的表现
用两种不同比例的肽制剂(酵母和大肠杆菌)(A和B,每种重复注射三次)加入人肽制剂中,以测定定量精确度。数据以1%q值处理(由软件自己报告;也就是说,DIA-NN和Spectronaut的有效FDR可能不同),使用DDA模式下对分组样品分析生成的谱图进行比对。混合物之间的肽比率通过LFQbench R包进行可视化(A:彩色虚线表示预期比率;橙色为酵母;绿色为人类;紫色为大肠杆菌)。图11B,Spectronaut和DIA-NN对肽和蛋白质的定量准确性如箱形图所示。
实验结论
在DIA-NN中引入的计算方法稳定且显著地增加了不同复杂度样品及不同质谱平台上获得定性和准确定量的肽和蛋白质的数量。DIA-NN首次通过使用短色谱梯度实现了蛋白质组的全面覆盖,从而显著缩短了质谱仪的运行时间,为以前无法实现的对高通量蛋白质组进行快速而精确的测量打开了大门。
推荐语
DIA模式是一种新的质谱数据采集方式。DIA技术将整个质谱全扫描范围分为若干个窗口,高速、循环地对每个窗口中所有离子进行碎裂和检测,无差异、无遗漏地获得样本中所有离子的全部碎片信息,并基于二级质谱定量。相对于传统的DDA模式其检测的重复性好,覆盖度高。
然而,DIA技术产生的数据非常复杂性。但通过DIA-NN软件的使用,可有效解决DIA数据繁杂难以处理的瓶颈,推动蛋白质组学的研究、发展。
众所周知,目前蛋白组学检测技术发展迅速,不仅iTRAQ/TMT技术能够对蛋白质进行定量检测,DIA作为蛋白组学新一代技术以及Nature Methods 关注技术,也越来越受蛋白组学研究科研者的青睐。
参考文献
1. Sun, S. et al. MS-Simulator: predicting y-ion intensities for peptides with two charges based on the intensity ratio of neighboring ions. J. Proteome Res. 11, 4509–4516 (2012).
2. Kingma, D. P . & Ba, J. Adam: A Method for Stochastic Optimization. Preprint at arXiv http://arxiv.org/abs/1412.6980 (2014).
3. Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 64, 479–498 (2002).
4. Röst, H. L. et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748 (2016).
5. Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
6. The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
7. Parker, S. J. et al. Indentification of a set of conserved eukaryotic internal retention time standards for data-independent acquisition mass spectrometry. Mol. Cell. Proteom. 14, 2800–2813 (2015).
8. Deutsch, E. W ., Lam, H. & Aebersold, R. PeptideAtlas: a resource for target selection for emerging targeting proteomics workflows. EMBO Rep. 9, 429–434 (2008).
9. Teleman, J. et al. DIANA—algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics 31, 555–562 (2015).
10. Mülleder, M., Campbell, K., Matsarskaia, O., Eckerstorfer, F . & Ralser, M. Saccharomyces cerevisiae single-copy plasmids for auxotrophy compensation, multiple marker selection, and for designing metabolically cooperating communities. F1000Res 5, 2351 (2016).
11. Perez-Riverol, Y . et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019)
END




